Registration No. 333-238240
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 4 to
FORM S-1
REGISTRATION STATEMENT UNDER THE SECURITIES ACT OF 1933
Propanc Biopharma, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 2834 | 33-0662986 | ||
(State of Incorporation) |
(Primary Standard Industrial Classification Number) |
(IRS Employer Identification Number) |
James Nathanielsz
Chief Executive Officer
Propanc Biopharma, Inc.
302, 6 Butler Street
Camberwell, VIC, 3124 Australia
+61-03-9882-6723
(Address, including zip code, and telephone number, including area code,
of registrant’s principal executive offices)
Please send copies of all communications to:
Lucosky Brookman LLP
101 Wood Avenue South, 5th Floor
Woodbridge, New Jersey 08830
Tel. No.: (732) 395-4400
Fax No.: (732) 395-4401
(Address, including zip code, and telephone, including area code)
Approximate date of proposed sale to the public:
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. [X]
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
If this Form is a post-effective amendment filed pursuant to rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer [ ] | Accelerated filer [ ] |
| Non-accelerated filer [X] | Smaller reporting company [X] |
| Emerging growth company [ ] |
If an emerging growth company, indicate by checkmark if the registrant has not elected to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. [ ]
CALCULATION OF REGISTRATION FEE
Title of Each Class of securities to be registered | Number of shares of common stock to be registered(1) | Proposed Maximum Offering Price Per Share | Proposed Maximum Aggregate Offering Price (2) | Amount
of Registration Fee | ||||||||||||
| Common Stock | 804,518 | $ | 0.04 | (3) | $ | 22,455 | $ | 2.91 | ||||||||
| Common Stock underlying Pre-funded Warrants to Purchase Common Stock (3) | 10,445,482 | $ | 0.04 | (3) | $ | 427,545 | $ | 55.50 | ||||||||
| Common Stock underlying Series A Warrants to Purchase Common Stock | 11,250,000 | $ | 0.20 | $ | 2,250,000 | $ | 292.05 | |||||||||
| Common Stock underlying Series B Warrants to Purchase Common Stock | 63,750,000 | $ | 0.04 | $ | 2,550,000 | $ | 330.99 | |||||||||
| Common Stock underlying Series C Warrants to Purchase Common Stock | 63,750,000 | $ | 0.20 | $ | 12,750,000 | $ | 1,654.95 | |||||||||
| Total | 150,000,000 | $ | 18,000,000 | (4) | $ | 2,336.40 | (5)(6) | |||||||||
| (1) | Includes up to an aggregate of 149,195,482 shares of the Company’s Common Stock, $0.001 par value per share (the “Common Stock”) issuable upon exercise of warrants that may be sold from time to time pursuant to this registration statement by the Selling Securityholder (as defined herein) identified herein. |
| (2) | Estimated solely for the purpose of computing the amount of the registration fee pursuant to Rule 457(o) under the Securities Act of 1933, as amended (the “Securities Act”). |
| (3) | Calculated based on the aggregate purchase price of $450,000 allocated ratably among the shares of common stock and pre-funded warrants to purchase stock issued at closing. |
| (4) | Pursuant to Rule 416 under the Securities Act, the shares of common stock registered hereby also include an indeterminate number of additional shares of common stock as may, from time to time, become issuable by reason of stock splits, stock dividends, recapitalizations or other similar transactions. |
| (5) | The fee is calculated by multiplying the aggregate offering amount by .0001298, pursuant to Section 6(b) of the Securities Act of 1933. |
(6) Previously paid.
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act or until the registration statement shall become effective on such date as the Commission, acting pursuant to said section 8(a), may determine.
PRELIMINARY PROSPECTUS SUBJECT TO COMPLETION DATED OCTOBER 13, 2020
The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting offers to buy these securities in any jurisdiction where the offer or sale is not permitted.
Propanc Biopharma, Inc.
150,000,000 Shares of Common Stock
This prospectus relates to the offering and resale by the Selling Stockholder identified herein of up to 150,000,000 shares of Common Stock of Propanc Biopharma, Inc. (the “Company”). These shares include Common Stock underlying the 7,500,000 units, with each unit consisting of (i) 1.5 shares of Common Stock, or pre-funded warrants (the “Pre-funded Warrants”) and (ii) 1.5 warrants to purchase one share of Common Stock (“Series A Warrants”, and collectively with the Common Stock the “Units”). Each Series A Warrant have an exercise price per share equal to $0.20 per share. The Series A Warrants will be exercisable immediately and will expire on the three-year anniversary of its original issuance date. The Purchaser is not permitted to exercise any portion of the Series A Warrant that would result in the Purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% of our outstanding common stock following the consummation of this offering.
The Purchaser is not permitted to exercise portion of the Pre-funded Warrant that would result in the Purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% of our outstanding common stock following the consummation of this offering. Each pre-funded warrant included in the pre-funded units will have a per share exercise price of $0.0001. The pre-funded warrants contained in the pre-funded units will be exercisable immediately and may be exercised at any time until the Pre-funded Warrants are exercised in full.
In addition to the Units, we are registering 63,750,000 shares of Common Stock underlying warrants (the “Series B Warrant”) currently outstanding. Each Series B Warrant has an exercise price per share equal to $0.04 per share and will expire on the three-year anniversary of its original issuance date. The Purchaser is not permitted to exercise portion of the Series B Warrants that would result in the Purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% of our outstanding common stock following the consummation of this offering.
We are also registering 63,750,000 shares of Common Stock underlying warrants (the “Series C Warrants” and together with the Series A Warrants, Pre-funded Warrants and Series B Warrants, the “Warrants”) that are currently outstanding. Each Series C Warrant has an exercise price per share equal to $0.20 per share. The Purchaser is not permitted to exercise portion of the Series C Warrants that would result in the Purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% of our outstanding common stock following the consummation of this offering. The Series C Warrants are not permitted to be exercised until the Series B Warrants are fully exercised.
There is no established public trading market for the Warrants, and we do not expect a market to develop. Without an active trading market, the liquidity of the Warrants will be limited. The actual offering price per Warrants will be negotiated between us and the Purchaser based on the trading of our common stock prior to the offering, among other things, and may be at a discount to the current market price. Therefore, the assumed public offering price used throughout this prospectus may not be indicative of the final offering price. Our shares of Common Stock are currently quoted on the OTCQB market operated by the OTC Markets Group, Inc., or “OTCQB,” under the ticker symbol “PPCB.” On October 6, 2020 the closing price as reported on the OTCQB was $0.0025 per share. This price will fluctuate based on the demand for our Common Stock.
The Selling Stockholder may offer all or part of the shares for resale from time to time through public or private transactions, at either prevailing market prices or at privately negotiated prices. See “Plan of Distribution” beginning on page 67 of this prospectus for more information.
We will not receive any of the proceeds from the sale of shares of common stock by the Selling Stockholder. See “Use of Proceeds.”
This prospectus provides a general description of the securities being offered. You should read this prospectus and the registration statement of which it forms a part before you invest in any securities.
Investing in our securities involves a high degree of risk. See “Risk Factors” beginning on page 40 of this prospectus for a discussion of information that should be considered in connection with an investment in our securities.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
You should rely only on the information contained in this prospectus or any prospectus supplement or amendment hereto. We have not authorized anyone to provide you with different information. If anyone provides you with different or inconsistent information, you should not rely upon it. This prospectus is not an offer to sell, nor is the selling stockholder seeking an offer to buy, securities in any state where such offer or solicitation is not permitted. The information in this prospectus is complete and accurate only as of the date on the front cover of this prospectus, regardless of the time of delivery of this prospectus or any sale of shares of our common stock. Our business, financial condition, results of operations and prospects may have changed since that date.
The date of this prospectus is October 13, 2020.
TABLE OF CONTENTS
Propanc Biopharma, Inc., the Propanc Biopharma logo, and other trademarks or service marks of Propanc Biopharma appearing in this prospectus are the property of Propanc Biopharma, Inc. This prospectus also includes trademarks, tradenames and service marks that are the property of other organizations. Solely for convenience, trademarks and tradenames referred to in this prospectus appear without the ® and ™ symbols, but those references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights, or that the applicable owner will not assert its rights, to these trademarks and tradenames.
You should rely only on the information contained in this prospectus or in any related free writing prospectus filed by us with the Securities and Exchange Commission (the “SEC”). We have not authorized anyone to provide you with any information or to make any representation not contained in this prospectus or incorporated by reference. We do not take any responsibility for, and can provide no assurance as to the reliability of, any information that others may provide to you. This prospectus is not an offer to sell or an offer to buy securities in any jurisdiction where offers and sales are not permitted. The information in this prospectus is accurate only as of its date, regardless of the time of delivery of this prospectus or any sale of securities. You should not assume that the information contained in this prospectus or any prospectus supplement or free writing prospectus is accurate as of any date other than the date on the front cover of those documents, or that the information contained in any document incorporated by reference is accurate as of any date other than the date of the document incorporated by reference, regardless of the time of delivery of this prospectus or any sale of a security. Our business, financial condition, results of operations and prospects may have changed since those dates.
We have not done anything that would permit a public offering of the securities or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of the securities and the distribution of this prospectus outside of the United States.
| 3 |
The following summary highlights information contained elsewhere or incorporated by reference in this prospectus and does not contain all of the information you should consider in making your investment decision. You should read this summary together with the more detailed information, including our consolidated financial statements and the related notes included in this prospectus, contained or incorporated by reference in this prospectus. You should carefully consider, among other things, the matters discussed under the headings “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and in our consolidated financial statements, before making an investment decision. You should also read and consider the information in the documents to which we have referred you in “Where You Can Find Additional Information” And “Incorporation of Certain Information by Reference.”
As used in this prospectus, unless the context otherwise requires, references to “we,” “us,” “our,” “our Company,” the “Company” and “Propanc” refer to Propanc Biopharma, Inc. and our wholly owned subsidiary Propanc PPY LTD.
Business Overview
Our Company
General
As used in this Registration Statement, references to the “Company,” “Propanc,” “we,” “our,” and “us” refer to Propanc Biopharma, Inc. and its consolidated subsidiary, unless otherwise indicated. In addition, references to our “financial statements” are to our consolidated financial statements included elsewhere in this Registration Statement except as the context otherwise requires.
We prepare our consolidated financial statements in United States dollars and in accordance with generally accepted accounting principles as applied in the United States, (“U.S. GAAP”). In this Registration Statement, references to “$” and “dollars” are to United States dollars.
Overview
We are a development-stage healthcare company that is currently focused on developing new cancer treatments for patients suffering from pancreatic, ovarian and colorectal cancer. Utilizing our scientific and oncology consultants, we have developed a rational, composite formulation of anti-cancer compounds, which together exert a number of effects designed to control or prevent tumors from recurring and spreading through the body. Our lead product candidate, PRP, is a variation upon our novel formulation and involves proenzymes, the inactive precursors of enzymes. As a result of positive early indications of the anti-cancer effects of our technology over the last 12-24 months we have conducted successful pre-clinical studies on PRP and also commenced preparation for a clinical study in advanced cancer patients. Subject to us receiving sufficient financing, we plan to begin our Investigational Medicinal Product Dossier, study proposal and Investigator’s Brochure in the second half of 2020 calendar year. Our plan is to then commence our study preparation process with the contract research organization, analytical lab and trial site(s) selection and to begin our clinical trial application for PRP (“CTA”) compilation in the third calendar quarter of 2020 and complete the CTA compilation and submit the CTA in the first calendar quarter of 2021. In first or second quarter of 2021, we plan to begin the preparation of logistics and trial site initiation visits. Subject to raising additional sufficient capital, we subsequently plan to commence a First-In-Human (FIH), Phase Ib study in patients with advanced solid tumors, evaluating the safety, pharmacokinetics and anti-tumor efficacy of PRP in the first half of 2021 calendar year, which study we hope to complete within twelve months thereafter. We intend to develop our PRP to treat early-stage cancer and pre-cancerous diseases and as a preventative measure for patients at risk of developing cancer based on genetic screening.
Key Research and Development Highlights:
| ● | Potential cancer treatment: We are developing PRP, an intravenous once-daily proenzyme treatment as a therapeutic option in cancer treatment and prevention. PRP is a combination of the pancreatic proenzymes trypsinogen and chymotrypsinogen. | |
| ● | Multiple mechanisms of action on cancerous or carcinogenic cells: PRP produces multiple effects on cancerous cells intended to inhibit tumor growth and potentially stop a tumor from spreading through the body. This is in contrast to current cancer treatments that lack sufficient efficacy to achieve a durable clinical response. As our research progresses, we intend to explore further these multiple mechanisms of action in order to identify opportunities to expand our intellectual property portfolio. Furthermore, we hope to uncover the molecular targets of the proenzymes to identify their potential for developing new compounds. |
| 4 |
| ● | Encouraging data from patient treatment: We began our development efforts by analyzing scientific research undertaken over the last 15 years, including clinical data from patients in the UK and Australia. We concluded that there is at least indirect evidence that a formulation such as PRP may be an effective treatment against cancer and warranted further development. | |
| ● | Pre-Clinical Efficacy Studies: In November 2015, we completed animal efficacy studies in mice through our contract research partner, vivoPharm, demonstrating proof of concept in vivo. During the course of these studies, we discovered a new target therapeutic dose range using proenzymes for treating cancer. That month, we filed a patent application in support of this discovery, as described further herein. | |
| ● | Pre-Clinical Toxicology Studies: In October 2016, we completed an animal study for PRP, in which we evaluated its toxicokinetic parameters as well as its distribution and bioavailability, both before and after repeat dosages. We then initiated a second such study in December 2016. That study escalated the dosage levels in different phases and was completed in April 2017. We observed no major toxicological findings after PRP was administered by intravenous injection once daily throughout the study period. | |
| ● | Anticipated Clinical Trial Application: With the successful completion of the studies described above, we believe we have accumulated sufficient data to establish a safe and effective dosage level for PRP and advance our product development to the clinical stage. We are currently working with our manufacturer to create the finished product that will be part of our Investigational Medicinal Product Dossier to be submitted in connection with our anticipated first clinical trial for PRP, which we expect will be conducted in the UK, Europe or Australia. | |
| ● | Orphan Drug Designation: In June 2017, we received notification from the U.S. Food and Drug Administration (FDA) that PRP had been conferred Orphan Drug Designation. This special status is granted when a rare disease or condition is implicated and a potential treatment qualifies under the Orphan Drug Act and applicable FDA regulations. Orphan Drug status qualifies us for various development incentives, including protocol assistance, the potential for research grants, the waiver of future application fees, and tax credits for clinical testing if we choose to host future clinical trials in the U.S. | |
| ● | Unique intellectual property: In addition to our pre-clinical studies, we have also focused on building a significant portfolio of intellectual property around the use of proenzymes in the treatment of cancer, identifying new formulations, alternative routes of administration and potential new therapeutic targets. We have filed numerous patent applications relating to PRP, several of which have been granted while others remain pending. In the U.S., we have been issued two patents to date (No. 9,636,359 and 10,350,239). A further application has been filed and an Examiner’s first report expected by May 31, 2022. Our patent protection extends to both PRP’s mechanism of action and the new compositions of proenzymes. |
| 5 |
| ● | Research and development expenses: During the fiscal years ended June 30, 2020 and 2019, we have spent $179,987 and $260,335, respectively, on research and development expenses. Historically, we have assumed all of the costs associated with research and development. In September 2018, the Company entered into a two-year collaboration agreement with the University of Jaén (the “University”) to provide certain research services to the Company. In consideration of such services, the Company agreed to pay the University approximately 52,000 Euros ($59,508 USD) in year one of which 31,754 Euros ($36,117 USD) was paid in fiscal 2019 and 15,410 Euros ($17,331 USD) was accrued in fiscal 2020; and a maximum of 40,000 Euros ($45,775 USD) in year two. Additionally, in exchange for full ownership of the intellectual property the Company agreed to pay royalties of 2% of net revenues to the University. |
In August 2019, we announced that we have developed a method to quantify the active ingredients of our lead product candidate, PRP, in preparation for the company’s First-In-Human (“FIH”) study, planned for the second half of 2021 calendar year. The work was conducted by Propanc’s research partner based in Berlin, Germany, who has extensive experience in the development of functional assays for unique bio-therapeutics. This bioanalytical method development and validation plays a significant role in evaluation and interpretation of the systemic absorption of PRP in clinical studies including its distribution, and clinical effects throughout the body. The development of the bioanalytical assay is also an important step for the clinical development of PRP, as Propanc evaluates sites to conduct the FIH study in advanced cancer patients, such as the Peter Mac Center, Australia’s largest cancer hospital, which has significant experience in early stage clinical development. Validation of the bioanalytical method will be undertaken in 2021.
In November 2019, we announced today that our POP1 research and drug discovery program has made significant advancements towards producing synthetic versions of the two proenzymes, trypsinogen and chymotrypsinogen. With the aim of producing large quantities of trypsinogen and chymotrypsinogen for commercial use, exhibiting minimal variation between lots and without sourcing the proenzymes from animals, the Company is undertaking a challenging research project in collaboration with the universities of Jaén and Granada. The two active ingredients are currently naturally derived from animal sources, which combine to form our lead product candidate, PRP. Our vision is to produce a backup product candidate to PRP which can further stabilize and enhance the effects of the proenzymes when administered to patients. At the research laboratories at the universities of Jaén and Granada, scientific researchers are in the process of optimizing conditions to achieve high titers of recombinant trypsinogen and chymotrypsinogen with this expression system.
In January 2020, we announced that a Certificate for Advance Overseas Finding was received from the Board of Innovation and Science Australia to receive up to a 43.5% “cash back” benefit from overseas R&D expenses. The finding relates to the planned Phase 1 clinical trial – Multiple Ascending Dose Studies of proteolytic proenzymes for the treatment of pancreatic cancer. Overseas activities to be undertaken include the development of an analytical assay for the quantification of active pharmaceutical ingredients in the Company’s lead product candidate, PRP, and its manufacture of the finished product for the Phase 1 clinical trial.
Company History
We were originally incorporated in Melbourne, Victoria Australia on October 15, 2007 as Propanc PTY LTD and continue to be based in Camberwell, Victoria Australia. Since our inception, substantially all of our operations have been focused on the development of new cancer treatments targeting high-risk patients, particularly cancer survivors, who need a follow-up, non-toxic, long-term therapy designed to prevent the cancer from returning and spreading. We anticipate establishing global markets for our products.
On November 23, 2010, our Company was incorporated in the state of Delaware as Propanc Health Group Corporation. In January 2011, to reorganize our Company, we acquired all of the outstanding shares of Propanc PTY LTD on a one-for-one basis and Propanc PPY LTD became our wholly-owned subsidiary. Effective April 20, 2017, we changed our name to “Propanc Biopharma, Inc.” to better reflect our stage of operations and development. On the same date, we also effected a 1-for-250 reverse stock split whereby we (i) decreased the number of authorized shares of our common stock to 100,000,000 (ii) decreased the number of authorized shares of our preferred stock to 1,500,005 and (iii) decreased, by a ratio of 1-for-250 the number of retroactively issued and outstanding shares of our common stock.
On January 23, 2018, we filed a Certificate of Amendment to our Certificate of Incorporation to increase the number of authorized shares of our common stock from 100,000,000 to 400,000,000. On September 21, 2018, we filed a Certificate of Amendment to our Certificate of Incorporation to increase the number of authorized shares of our common stock from 400,000,000 to 4,000,000,000. On June 11, 2019, we filed a Certificate of Amendment, as amended, to our Certificate of Incorporation to decrease the number of authorized shares of our common stock from 4,000,000,000 to 100,000,000 in connection with the 1-for-500 reverse stock split that occurred on June 24, 2019. On March 15, 2020, we filed a Certificate of Amendment, as amended, to our Certificate of Incorporation to increase the number of authorized shares of our common stock from 100,000,000 to 1,000,000,000.
On February 4, 2020, we filed a Certificate of Amendment to our Certificate of Incorporation to increase the number of authorized shares of our common stock from 100,000,000 to 1,000,000,000 which was effected on March 13, 2020.
Important Milestones for Propanc
| ● | From the late 1990s, work from other scientists and clinicians, including Dr. Josef Novak in the U.S., and a since retired oncologist from the Czech Republic, Dr. Frantisek Trnka, shed new light on the therapeutic potential of Professor John Beard’s insights. Extensive laboratory work undertaken over a number of years by Novak and Trnka was reported in the journal Anticancer Research in 2005 in the paper entitled Proenzyme Therapy of Cancer. The conclusion of Novak and Trnka from this work was the discovery “that proenzyme therapy mandated first by John Beard nearly one hundred years ago, shows remarkable selective effects that result in growth inhibition of tumor cells with metastatic potential.” Today, these important scientific observations support our view that proenzymes are selective and effective in targeting malignant tumor cells and could become an effective tool in the fight against metastatic cancer. |
| 6 |
| ● | In 2007, Dr. Julian Kenyon, Medical Director of the Dove Clinic in the UK, and Dr. Douglas Mitchell further developed the therapeutic concepts of Beard and identified strategies that could improve upon the therapeutic potential of Beard’s original ground-breaking work. A suppository formulation was developed by Mandeville Medicines in Buckinghamshire, UK, at the request of, and in consultation with, Drs. Kenyon and Mitchell, in an effort to improve on results reported in the literature pertaining to the potential therapeutic use of proenzymes in cancer treatment. Patients were first treated with the suppository formulation in April 2007 at The Dove Clinic in the UK, and in July 2007 at the Opal Clinic in Australia. Drs. Kenyon and Mitchell, through The Dove Clinic and Opal Clinic respectively, treated cancer patients in the United Kingdom and Australia with a suppository formulation of proenzymes. The treatment was undertaken under special UK and Australian regulatory provisions. In the UK it was undertaken under the regulations of the Medicines and Healthcare Products Regulatory Agency (the “MHRA”), designed for patients who have special clinical needs that cannot be met by licensed medicinal products, and in Australia under the Therapeutic Goods Administration (“TGA”) Special Access Scheme, a mechanism that provides for the import and/or supply of an unapproved therapeutic good for a single patient, on a case by case basis. In both jurisdictions, patients are permitted to receive treatment on an individual basis for compassionate use as long it is supplied by a recognized, licensed manufacturer who is able to meet certain guidelines for unapproved products, and individual case files are maintained for patients should the regulatory authorities require this information. No prior approval was required by either the MHRA or TGA prior to the commencement of treatment. No suppository formulation of the proenzymes was available and it was necessary for a novel suppository formulation to be manufactured specifically for these patients by a suitably licensed manufacturer. | |
| ● | Forty-six late stage cancer patients suffering from a range of malignancies in the UK and Australia received treatment with the proenzyme suppositories over periods of time ranging from one month to in excess of 17 months. Inspired by their observations in clinical practice, Drs. Kenyon and Mitchell resolved to develop proenzyme therapy for cancer patients worldwide. | |
| ● | In late 2007, Drs. Kenyon and Mitchell and Mr. James Nathanielsz, our Chief Executive Officer and Chief Financial Officer, developed a strategy to commercialize the newly developed proenzyme formulation, now designated PRP. Propanc PTY LTD. was established in Australia as a vehicle to refine, develop and commercialize novel, patented proenzyme therapeutics for the treatment of cancer. | |
| ● | In 2008, our Scientific Advisory Board (the “Scientific Advisory Board”) comprising Professor John Smyth (Edinburgh University), Professor Klaus Kutz (Bonn University) and Professor Karrar Khan (De Montfort University) was established. Today, the expertise of the Scientific Advisory Board in oncology research and development will be relied upon as we initiate patient trials and advance our products down the requisite regulatory pathways to commercialize our proenzyme therapies. | |
| ● | In 2009, a retrospective review of the patient notes from the 46 patients treated in the UK and Australia with the proenzymes suppositories (as described above) was undertaken by Dr. Kenyon. This report was subject to analysis by Professor Klaus Kutz who, at the time of the review, was an independent consultant in clinical pharmacology and safety, specializing in oncology. Professor Kutz observed that no patients were reported as living for a period less than that predicted by the treating clinician and a number of terminally ill patients lived marginally longer than predicted, particularly those suffering from pancreatic, colorectal, ovarian and gastro-intestinal cancers. As a result of the observations made by Dr. Kenyon and Professor Kutz, we are targeting the development of proenzyme therapy for the treatment of colorectal and pancreatic cancers for clinical trials, and in the future targeting other cancer types as our product candidate progresses to commercialization. |
| 7 |
| ● | In early 2008, a research collaborative partnership was established with Professor David Tosh at the Center for Regenerative Medicine, Department of Biology and Biochemistry at Bath University, to investigate the molecular mechanisms by which the proenzyme formulation is acting, which resulted in us filing two provisional patents a year later. We undertook additional scientific research with Professor Tosh, Dr. Macarena Perán, Department of Health Sciences at Jaén University, and Dr. Juan Antonio Marchal, Biopathology and Regenerative Medicine Institute at Granada University. Important anti-cancer effects of the proenzymes were discovered, including triggering cell necrosis (cell death) and apoptosis (programmed cell death) and significantly, the induction of cell differentiation (i.e. inducing cancer cells to exhibit normal cell behavior). This led to us increasing our intellectual property base and patent new pharmaceutical compositions designed to enhance the effects of proenzymes. Subsequently, two provisional patents were combined into one Patent Cooperation Treaty (“PCT”) Application, filed on October 22, 2010 (PCT Application), and then a year later, we completed a 30 month national phase filing deadline for an international patent and commenced entering the national phase in countries around the world. Thus far, we have received grant status in Australia, China, Japan, Indonesia, Israel, New Zealand, Singapore and South Africa and our application remains under examination in Brazil, Canada, the European Union, Malaysia, Mexico and the Republic of Korea. In the United States, two patents have been issued to date by the United States Patent and Trademark Office (No. 9,636,359 and No. 10,350,239) while another remains pending. We also have another 3 PCT applications for proenzyme compositions that have entered national phase in major global jurisdictions. | |
| ● | In late 2010, we made important discoveries and scientific observations, resulting in additional composition claims, which were included in the original PCT Application, further protecting the company’s proenzyme formulation. Collaboration with vivoPharm Pty Ltd. (“vivoPharm”), located in Melbourne, Australia, with research facilities in Hershey, Pennsylvania, United States, identified a highly synergistic ratio of the proenzymes when combined together, resulting in increased anti-cancer effects in several tumor cell lines. Furthermore, although α-Amylase was previously included in the early days of enzyme therapy and in the suppository formulation developed by Dr. Kenyon and Dr. Mitchell, after evaluating the synergistic interaction between the two proenzymes and α-Amylase, we concluded that α-Amylase did not contribute to the anti-tumor activity of the formulation, and so it was removed. By 2011, further work completed by vivoPharm confirmed the anti-metastatic effects of the newly combined ratio of the proenzymes in various cell line assays, and anti-angiogenic (inhibition of blood vessel formation) properties of the proenzyme treatment in mice. | |
| ● | At this time, we decided to access the U.S. markets in order to raise the capital needed to finance the Company’s proenzyme treatment for future preclinical testing and clinical trials. We incorporated as Propanc Health Group Corporation in the state of Delaware in November 2010 and in January 2011, we acquired all of the outstanding shares of Propanc PTY LTD on a one-for-one basis making and Propanc PTY LTD became our wholly-owned subsidiary. In mid-2012, our common stock began trading on the Over-the-Counter Bulletin Board and it currently trades on OTCQB.
| |
| ● | In May 2013, it was observed that proenzymes enforce the re-entry of cancer cells back into normal cellular pathways and this may represent a novel approach to the treatment of cancer. These findings were published in Cellular Oncology, a peer reviewed journal of the International Society for Cellular Oncology. | |
| ● | In 2014, after conducting a detailed strategic review of our scientific and preclinical research, our development team determined that parenteral drug administration is the preferred route for the Company’s lead product, PRP. This approach is expected to maximize results in future patient trials, by ensuring maximum exposure of the drug to the tumor site. |
| 8 |
| ● | In mid-2015, Dr. Joseph Chalil joined our Scientific Advisory Board as an independent expert to provide advice on the Company’s drug development programs, in particular, our lead product, PRP. Dr. Chalil is a physician and executive at Boehringer Ingelheim, one of the world’s largest privately held pharmaceutical companies. | |
| ● | Between July 2015 and February 2016, several scientific research findings were announced demonstrating significant anti-tumor efficacy in several animal models, including pancreatic and ovarian cancers at higher doses when administering proenzymes by intravenous injection, dramatic suppression of cancer stems cells in cell culture by altering several key pathways involved with invasion and metastasis, and identification of a synergistic response in a broad range of cancer types including kidney, melanoma, brain, prostate, liver, uterine and lung cancers. | |
| ● | In 2016, we added additional members from our partner universities and hospital to our Scientific Advisory Board, including Dr. Macarena Perán, who is currently Reader in Anatomy at the University of Jaén in Spain, Professor Juan Antonio Marchal Corrales, Professor of Anatomy and Embryology at the Faculty of Medicine at the University of Granada, and Dr. Maria García, Head of Translational Research at the University Hospital of Granada. | |
| ● | In August 2016, we entered into a Manufacturing Services Agreement and Quality Assurance Agreement with Amatsigroup NV, formally known as Q-Biologicals NV, a contract manufacturing organization located in Belgium. Pursuant to the Manufacturing Services Agreement, Amatsigroup produces for us certain drug substances and product containing certain enzymes at its facility in Belgium. We use these substances and products for development purposes, including but not limited to future clinical trials. | |
| ● | In October 2016, we completed an animal study for PRP, in which we evaluated its toxicokinetic parameters as well as its distribution and bioavailability, both before and after repeat dosages. We then initiated a second such study in December 2016. That study escalated the dosage levels in different phases and was completed in April 2017. We observed no major toxicological findings after PRP was administered by intravenous injection once daily throughout the study period. | |
| ● | On April 20, 2017, we changed our corporate name to “Propanc Biopharma, Inc.” to better reflect our stage of operations and development. | |
| ● | In June 2017, we received notification from the FDA that PRP had been granted Orphan Drug Designation, a special status that will enable us to qualify for tax credits for our future clinical trials, among other benefits. | |
| ● | In October 2017, we published key findings relating to a combination of two proenzymes trypsinogen and chymotrypsinogen A with potent in vitro and in vivo anti-tumor efficacy in Scientific Reports, a peer reviewed scientific journal covering all areas of the natural sciences. It was concluded that PRP could have relevant oncological clinical applications for the treatment of advanced or metastatic adenocarcinoma and advanced epithelial ovarian cancer. |
| 9 |
| ● | In February 2018, we announced allowance of our key patent application from the European Patent Office (EPO) covering a pharmaceutical composition for treating cancer comprising trypsinogen and chymotrypsinogen within the European Union. The allowed patent application is the first approval for the Company in the EU, which protects the Company’s lead product candidate, PRP, a solution for once-daily intravenous administration of a combination of two pancreatic proenzymes trypsinogen and chymotrypsinogen. | |
| ● | In March 2018, we completed the successful reproduction run of the manufacturing process for the Company’s two drug substances trypsinogen and chymotrypsinogen. The successful reproduction run demonstrated scalability of our proprietary manufacturing process to enable routine production of the two active substances for PRP. The process was developed in collaboration with a European Contract Manufacturing Organization (CMO) experienced in the production of biopharmaceuticals. | |
| ● | In July 2018, we entered national phase for two of our key patent applications from our intellectual property portfolio. The first patent application, which entered national phase in July 2018, describes a method to eradicate cancer stem cells, and a second patent application, covering proenzyme compositions for the treatment of solid tumors, completed national phase entry mid-July 2018. National phase is a process whereby applicants file a patent application in each individual jurisdiction or country, according to where intellectual property protection is sought. | |
| ● | In September 2018, we entered into a two-year collaboration agreement with the University of Jaén to provide certain research services to us. In consideration of such services, we agreed to pay the university approximately 52,000 Euros ($59,508 USD) in year one and a maximum of 40,000 Euros ($45,775 USD) in year two. Additionally, in exchange for full ownership of the intellectual property we agreed to pay royalties of 2% of net revenues to the University. | |
| ● | In December 2018, we announced that our foundation patent application has been granted by the Office of the Controller General of Patents, Design and Trademarks, India. The foundation patent, which covers our lead product candidate, PRP, pioneers the discovery of a pharmaceutical composition for treating cancer via a combination of trypsinogen and/or chymotrypsinogen pancreatic proenzymes. As of June 30, 2019, the foundation patent has been granted in the United States, Belgium, Czech Republic, Denmark, France, Germany, Ireland, Italy, Netherlands, Portugal, Spain, Sweden, Switzerland, Liechtenstein, Turkey, United Kingdom, Australia, China, Japan, Indonesia, Israel, New Zealand, Singapore, Malaysia, South Africa, Mexico, Republic of Korea and India. It is presently under examination in Brazil and Canada. |
| 10 |
| ● | In January 2019, we announced that a cooperation agreement has been entered into between the University of Jaén and our Company to commence the POP1 joint drug discovery program to be co-funded by both parties. The agreement coincides with the appointment of research scientist, Mr. Aitor González, to lead the drug discovery and research activities over the next 3 to 4 years. The objective of the program is to identify and develop suitable backup compounds to our lead product candidate, PRP. As part of the agreement, Macarena Perán, Ph.D. and Julian Kenyon, M.D. have been appointed as joint supervisors, representing the University and our Company, respectively. The program involves advancing new compounds through a drug screening process, followed by preclinical and early stage clinical development. As the drug candidate progresses along the development pathway, the collaboration will also involve the Universities of Granada and Jaén, as well as Granada and Almeria Hospitals, which are members of FIBAO, a Public Health Foundation, based in Granada, Spain, committed to assisting commercial partners with the development and commercialization of innovative technologies designed to benefit humankind. | |
| ● | In March 2019, we announced that we received a notification of allowance from the United States Patent and Trademark Office (“USPTO”) confirming both methods of treatment and composition of matter claims involving trypsinogen and chymotrypsinogen for our foundation patent in the U.S. that covers PRP. The notification of allowance from the USPTO signifies that a USPTO examiner has determined that PRP’s patent application is complete and meets all relevant statutory requirements. | |
| ● | In March 2019, we announced that we have initiated development of a bio-analytical assay intended to quantify the active ingredients of RPR in preparation for human trails planned for the beginning of 2020 calendar year. The work will be conducted by a specialist Contract Research Organization with extensive knowledge in the development of functional assays for different bio-therapeutics. | |
| ● | In August 2019, we published a third scientific paper in a peer reviewed journal, Scientific Reports, highlighting the mechanism of action of proenzymes and its anti-cancer effects against cancer stem cells. In particular, we proved that proenzymes impaired engrafting of human derived pancreatic cancer stem cells in immune compromised mice, whilst also displaying an anti-growth effect towards grafted, human derived tumors. Our scientific researchers concluded that as cancer treatment moves towards more personalized medicine, proven therapies that target and treat specifically cancer stem cells may prove to be a useful method for reducing recurrence after drug treatment failures. | |
| ● | In November 2019, we made significant advancements through our POP1 joint research and drug discovery program, by producing synthetic recombinant versions of the two proenzymes, trypsinogen and chymotrypsinogen. The two active ingredients are currently naturally derived from animal sources, which combine to form PRP. Our vision is to further stabilize and enhance the effects of the proenzymes when administered to patients. Our scientific researchers have developed a novel expression system and are in the process of optimizing conditions to achieve high titers of recombinant trypsinogen and chymotrypsinogen. | |
| ● | In January 2020, we received a Certificate for Advance Overseas Finding from the Board of Innovation and Science Australia to receive up to a 43.5% “cash back” benefit from overseas R&D expenses. Overseas activities to be undertaken include the development of an analytical assay for the quantification of active pharmaceutical ingredients in PRP and its manufacture of the finished product for a Phase I clinical trial. | |
| ● | Today, after deepening our scientific knowledge of the anti-cancer effects of proenzymes through our ongoing efforts with our research partners and strengthening our intellectual property portfolio by filing our patents in countries around the world, we believe we are ready to undertake human clinical trials and subject to receiving adequate financing, we hope to submit a clinical trial application in the second quarter of 2021 calendar year. |
The Problem
In the early phases of tumor progression, cancer cells multiply near the site where their predecessors first began uncontrolled proliferation. The result, usually over a long period of time, is a primary tumor mass. Tumors often need to reach a large size before they make themselves apparent to the individual concerned, or the clinician screening for them.
Eventually, tumors of substantial size may begin to compromise the functioning of organs in which they have arisen and begin to evoke symptoms. In many cases, the effects on normal tissue function come from the physical pressure exerted by the expanding tumor masses. For example, large tumors in the colon may obstruct digestion products through the lumen, or in the lungs, airways may be compromised.
As dangerous and threatening as these primary tumors are, they are ultimately responsible for only about 10% of deaths. A far greater threat often arises for the patient, even after a primary tumor has been identified and removed. This threat involves cancerous growths that are discovered at sites far removed from the locations in their bodies where their primary tumors first appeared. These cancerous growths, called metastases, are responsible for approximately 90% of patient deaths from cancer. Metastases are formed by cancer cells that have left the primary tumor mass and traveled by the body’s blood and lymphatic vessels (a vein-like vessel carrying lymph, or white blood cells, from the tissues) to seek new sites and form new colonies. For example, breast cancers often spawn metastatic colonies in many tissues throughout the body including the brain, liver, bones, and lungs.
| 11 |
For primary tumors that have not yet metastasized, current treatments for cancer can be effective in initially reducing tumor burden. However, for many forms of cancer, current treatments lack sufficient efficacy to achieve a long lasting clinical response. Therefore, a vast majority of patients who succumb to cancer are killed by tumors that have metastasized. According to the National Cancer Institute’s SEER Cancer Statistics Review (2001 – 2007), of the patients diagnosed with late stage metastatic breast cancer, only 23% are expected to live longer than five years. This is compared to a 98% five-year survival rate for an early stage breast cancer patient when the cancer is confined to the primary site.
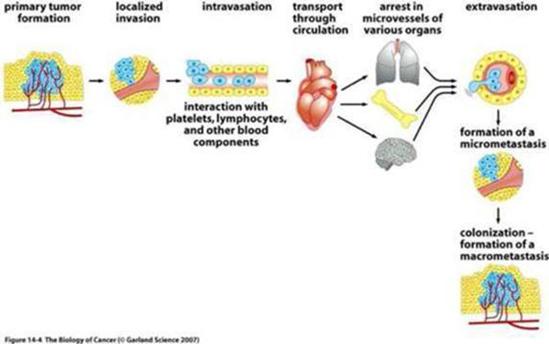
The invasion-metastasis cascade
The great majority of life threatening cancers occur in epithelial tissues, yielding carcinomas. Epithelial cells generally have a multi-sided, uniform shape. They have well defined contact points with neighboring cells and a strong attachment to the underlying connective tissue, or stroma, which creates a framework for solid tumors in the body. Separating the two is the specialized type of extracellular matrix, known as the basement membrane.
By definition, carcinomas that originate on the epithelial side of the basement membrane are considered to be benign; as long as the cells forming them remain on the same side. However, many carcinomas acquire the ability to penetrate the basement membrane, and individual cancer cells or groups of cancer cells begin to invade the stroma. This mass of cells is now reclassified as malignant. Often, many pathologists and surgeons reserve the label “cancer” for those epithelial tumors that have acquired this invasive ability.
Thereafter, carcinoma cells may invade into lymphatic or blood microvessels. The latter may then transport these cancer cells to distant sites in the body where they may be trapped and subsequently form new metastases.
It is important to note, that even before cells penetrate the basement membrane, they often stimulate angiogenesis (blood vessel formation) on the stromal side of the membrane, by expressing angiogenic proteins through the porous barrier. Not only does this enhance the ability of malignant cells to circulate into the blood, but also provides an important feedback loop for the cancer cell to maintain its invasiveness.
| 12 |
Understanding the mechanism by which benign cells change to a malignant state is therefore pivotal to developing anti-cancer treatments that have sufficient efficacy to achieve a long lasting clinical response.
The epithelial-mesenchymal transition and associated loss of E-cadherin expression enable carcinoma cells to become invasive.
Epithelial cells can undergo a transformation to a different cell type, called mesenchymal cells, through a process called the epithelial-to-mesenchymal transition (“EMT”). Mesenchymal cells have an elongated spindle shape, lack orderly contacts with neighboring cells and can survive without contact with a surface or connective tissue. The EMT process is a series of events that normally occur during the development of tissues and organs prior to birth, and also apply to normal wound healing processes. However, the same EMT process can also be applied to epithelial cancer cells, or carcinomas. When epithelial carcinoma cells residing in a solid tumor undergo the EMT process, the resulting mesenchymal cancer cells can invade through local barriers and metastasize to other parts of the body.
In addition to becoming invasive and motile after undergoing the EMT process, the resulting mesenchymal cells have significantly increased resistance to current cancer treatments. For example, in Cancer Research in 2005, it was reported that lung cancer cells expressing mesenchymal biomarkers appeared to be resistant to Tarceva and other targeted anti-cancer agents when transplanted into mice.
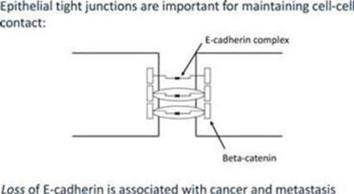
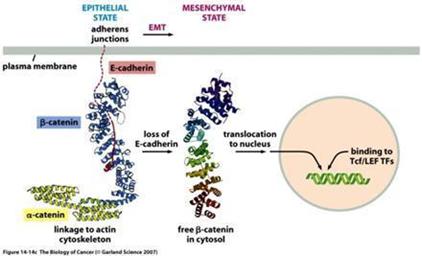
At the center of this critical process for transforming benign cells into carcinomas, is the protein Epithelial Cadherin (“E-Cadherin”). In normal cells, E-cadherin is located in the membrane and involved in maintaining cell to cell contact, which is critical to normal function and structure of epithelial tissues. The individual E-Cadherin molecules are attached to the actin (scaffolding, or cytoskeleton structure) within the cell, anchored by β-catenin, a protein which helps form the junction between epithelial cells. As well as forming an anchor between epithelial cells, β-catenin is also involved in gene transcription, a process by which DNA (deoxyribose nucleic acid) is converted into RNA (ribose nucleic acid) within the nucleus of a cell for the purpose of producing new proteins normally associated with routine cell function.
In the case of tumors, when cells become invasive, E-Cadherin expression decreases substantially, and β-catenin becomes free within the cell, which may then migrate to the nucleus and induce expression of the EMT program. Furthermore, once cells undergo an EMT, they begin to produce their own cytokines (cell signaling molecules), such as Transforming Growth Factor β, (“TGF-β”). This protein plays a critical multi-functional role in promoting angiogenesis, immunosuppression (suppressing the immune system from recognizing and attacking cancer cells), and maintaining their mesenchymal cell structure for prolonged periods via a feedback mechanism. Studies also suggest that TGF-β works with β-catenin to cause epithelial cancer cells to undergo an EMT.
| 13 |
A study in the British Journal of Cancer in 2011 demonstrated that in cholangiocarcinoma (bile duct cancer) cell lines, treatment of TGF-β increased cell migration, invasion and mesenchymal changes. Furthermore, expression of E-cadherin and N-cadherin was measured from resected (cut out) specimens from extra-hepatic (outside the liver) cholangiocarcinoma patients. Patients with low E-cadherin expression had a significantly lower survival rate than patients with high E-cadherin expression. They concluded the cadherin switch via TGF-β induced EMT in extra-hepatic cholangiocarcinoma leads to cancer progression.
Conversely, in studies of several types of carcinoma cells that had lost E-cadherin expression, re-expression of this protein strongly suppressed the invasiveness and motility of these cancer cells.
Together, these observations indicate that E-Cadherin levels is a key determinant of the biological behavior of epithelial cancer cells and that the cell to cell contact constructed by E-cadherin molecules impede invasiveness and hence metastasis.
Our Solution
Our solution is to develop and commercialize a long-term therapy to prevent tumor recurrence and metastases, the main cause of patient death from cancer. We believe this problem can be addressed by developing a proenzyme formulation specifically targeting malignant carcinoma cells to create a long lasting clinical benefit to the patient.
Propanc’s Theory Proenzymes Regulate Cell Proliferation
More than 100 years ago, Professor John Beard, a comparative embryologist, made an observation that the pancreas develops in most vertebrates at the time when the placenta begins to slow its rate of growth. He hypothesized that enzymes produced by the developing pancreatic gland curtail trophoblastic invasion (a rare condition in which abnormal cells grow inside the uterus from tissue that forms after conception) and suggested that pancreatic extracts should have a similar inhibitory effect on invasive tumors.
Subsequently in the late 1990s, after following Professor Beard’s recommendations, Drs. Novak and Trnka hypothesized that administration of proenzymes, rather than the enzymes, was of crucial importance to the clinical effectiveness of the treatment approach first developed by Professor Beard, and that the precursor nature of the active enzymes may offer protection against numerous serpins (proteins which can inhibit proenzymes) in the blood.
| 14 |
As knowledge of tumor cell and molecular cell biology has increased over the years, our scientists and research partners have made important scientific discoveries identifying that proenzymes suppress the EMT program and induce cell differentiation, i.e., return cancerous cells towards normal cell behavior, or a benign state.
After more than 100 years, the initial observations made by Professor Beard may have a potential common link between embryogenesis and cancer, by which cells are able to become motile and invasive, via the EMT program, where the administration of proenzymes may regulate cell proliferation as a means to controlling carcinomas.
PRP
Our lead product, PRP, is a novel, patented formulation consisting of two proenzymes, trypsinogen and chymotrypsinogen, combined at a ratio of one-to-six (1:6), to be administered intravenously. After establishing proof of concept in vivo as described earlier, supplemented by laboratory research at the Universities of Jaén and Granada on the mechanism of action of the proenzyme mixture, evidence suggests PRP may be effective against a range of solid tumors.
Selectivity
Research published by Novak and Trnka in Anticancer Research (2005) suggests that the proenzymes in our product, trypsinogen and chymotrypsinogen, exhibit specificity for tumor cells and not normal cells. Once activated, they in turn activate Protease Activated Receptors Type 2 (“PAR2”), which are located on the cell membrane and involved with cancer cell proliferation. Activation of PAR2 results in a cascade of intracellular activities, including activation of a major component of the cell which controls its structure and architecture, the actin cytoskeleton. In a cancer cell, proenzymes have the effect of converting globular actin into filamentous actin, which causes the cell structure to collapse and induce cell death. This reduces tumor volume and is often seen in clinical practice.
Anti-Cancer Effects and Mechanism of Action
PRP consists of proenzymes which are known to influence a number of pathways critical for cancer cells to invade, grow and metastasize. Research published in collaboration with our research partners at Jaén and Granada Universities in the Journal of Cellular Oncology in 2013 shows the clinical benefits of PRP appear to result from enhanced differentiation of tumor cells, which inhibits proliferation and consequently reduces their ability to invade and metastasize.
Specifically, the research showed that proenzymes:
| ● | induce a dose-dependent inhibition of cell growth, triggering apoptosis and cell necrosis; | |
| ● | enhance expression of epithelial markers, such as E-cadherin and β-catenin; | |
| ● | decrease expression of EMT transcription factors responsible for coding specific gene sequences from DNA, associated with TGF-β cell signaling pathways; and | |
| ● | induce malignant cells to differentiate to benign forms. |
Once activated, proenzymes influence the micro-immune environment around the cell, altering a number of pathways critical for supporting cancer cell growth, invasion and metastasis. This includes interacting with proteinases and cell signaling pathways in the extracellular matrix, whilst also interacting directly with cell surface proteins that effect the internal pathways of the cancer cell, triggering re-expression of epithelial markers, reducing important EMT markers, and inducing a series of cellular activities which alters the cancer cell’s morphology (structure) from a malignant to a benign state. Up to four pathways related to cancer spread and metastasis, including TGFΒ, Hippo, Wnt and Notch pathways were regulated by proenzymes.
| 15 |
Planned Clinical Development
PRP recently completed preclinical development. A First-In-Human (FIH), Phase Ib study in patients with advanced solid tumors, evaluating the safety, pharmacokinetics and anti-tumor efficacy of PRP is planned to commence in the second half of 2020 calendar year in a jurisdiction to be selected by us, subject to us receiving adequate financing, and is hoped to be completed within twelve months. The study will be an open-label, multicenter, non-comparative study of PRP administered at increasing dose levels, with once daily intravenous injections over a 28-day cycle, with at least 20 and up to 40 patients enrolled.
The Phase Ib study is planned to be followed by two open Phase IIa studies evaluating the safety, pharmacokinetics and anti-tumor efficacy of PRP administered intravenously to patients with locally advanced or metastatic pancreatic adenocarcinoma, or to patients with advanced epithelial ovarian cancer who have failed prior anti-cancer therapy regimen. These studies are envisioned to start in parallel, shortly after the FIH Phase IIa study, and are hoped to be finalized in 2022. Both studies will be open, multicenter phase II studies measuring overall survival of patients having received once daily intravenous administrations of PRP.
Preclinical Development
We have extensive in vitro and in vivo studies demonstrating the anti-tumor efficacy of a novel proenzyme formulation consisting of a combination of trypsinogen and chymotrypsinogen in a synergistic ratio. The preclinical work was undertaken in collaboration with our contract research organization, vivoPharm, in both Melbourne, Australia and Hummelstown, PA, United States, together with universities we partnered with, including the Biopathology and Regenerative Medicine Institute, Center for Biomedical Research, at the University of Granada in Granada, Spain, and the Department of Health Sciences at the University of Jaén in Jaén, Spain. We funded both vivoPharm and the universities to carry out this research and retained the intellectual property rights within the field relating to any discoveries based on the mechanism of action and anti-tumor effects of the proenzymes.
The following preclinical development activities have been undertaken to date:
| ● | We tested the anti-proliferative effects of trypsinogen and chymotrypsinogen in 24 cancer cell lines and determined a synergistic ratio of 1:6, which we used to formulate PRP; | |
| ● | We evaluated the in vitro anti-angiogenic effects of PRP, by soft-agar formation assay, and in vivo using the AngioChamber™ assay, which is based on the normal physiological process of wound healing, to promote fibrous capsule formation around an implanted growth factor-releasing Teflon chamber; | |
| ● | To analyze the anti-metastatic effects of proenzymes, we studied the effects of PRP in cell invasion, cell migration, and in the modulation of EMT related genes in pancreatic and ovarian cancer cells; and | |
| ● | We also performed in vivo a pharmacokinetic study and assessed the anti-tumor efficacy of PRP in murine cancer models. To accomplish this, we treated mice that were orthotopically inoculated with A2780 human ovarian cancer cells, or with Pan02 mouse pancreatic tumor cells, with PRP. |
| 16 |
Determination of Optimal Proenzyme Ratio
In this study, we determined first the half-maximal inhibitory concentrations (IC50) trypsinogen and chymotrypsinogen to measure their effect as single test articles in an extended panel of 24 human cancer cell lines. The IC50 values of trypsinogen ranged from 2.5 to 17.5 mg/ml and from 1.4 to 25.2 mg/ml for chymotrypsinogen. The IC50 values of trypsinogen were the basis for the calculation of concentration ratios for the combination of trypsinogen and chymotrypsinogen at 1:1, 1:2, 1:4, 1:6, 1:8, and 1:10. At these ratios, the growth inhibitory properties of the combination were evaluated in 24 cancer cell lines. Based on the coefficient of drug interaction (CDI) values, the combination of trypsinogen and chymotrypsinogen demonstrated greater growth inhibition at ratios of 1:4, 1:6, and 1:8, compared to the 1:1 ratio in most cell lines tested. Finally, a ratio of trypsinogen to chymotrypsinogen of 1:6 was determined to be the optimal formulation and used for later experiments.
Determination of the Coefficient of Drug Interaction
The representative graphs show an optimal pro-enzyme synergistic ratio of the Trypsinogen to Chymotrypsinogen as 1:6
| 17 |
Anti-angiogenic efficacy of pancreatic proenzyme formulation
To determine whether PRP affects angiogenesis, we used a soft-agar tube formation assay. Dispersed human umbilical vein endothelial cells (HUVEC) organized into clusters after three hours and began to form tube-like structures after five hours that were clearly evident after 24 hours. In contrast, PRP treated HUVECs presented a marked reduction in the number and length of closed capillary tubes in a concentration dependent manner, with a total disappearance of the structures after treatment with trypsinogen to chymotrypsinogen (T/C) 0.07/0.42 mg/ml.
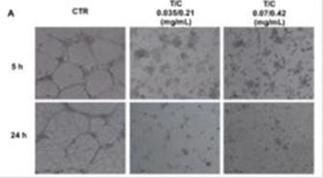
To assess if the inhibition of the tubule-like structures formation could be due to cell death caused by PRP treatment, CellTracker Green/CMFDA staining was used to identify viable cells. Both control and PRP treated cells showed green staining, indicating that the inhibition of cellular cords was independent from cell viability.
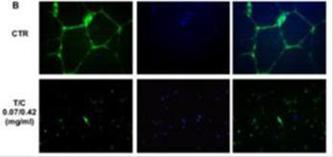
Furthermore, quantification of the number of capillary-like structures at different areas of the cell revealed a dramatic and significant difference between the number of structures formed by non-treated cells when compared with PRP-treated cells (p <0.01 vs. Control).
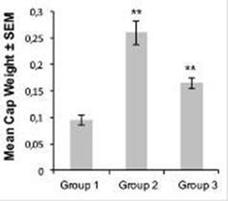
The anti-angiogenic effect of PRP was additionally investigated in vivo using the AngioChamber™ assay, a model used to assess the efficacy of anti-angiogenic treatments by measuring fibrous capsule formation in mice. In this assay the inclusion of basic fibroblast growth factor (bFGF) in the chamber supports the induction of blood vessels development and formation of a fibrous capsule. AngioChamber™ were excised from all post-mortem mice on the termination day, 24 hours following final treatment (Day 5).
The results show that fibrous capsule formation was significantly greater in the vehicle control group with bFGF captured in the chamber (Group 2, Induction Control) than in the vehicle control group without bFGF loaded into the chamber (Group 1, Baseline Control) (p<0.05) indicating that bFGF adequately and significantly stimulated capsule formation. Furthermore, treatment with PRP (Group 3) resulted in a significant reduction in angiogenesis compared to the induction control (Group 2), as indicated by the difference in capsule weight (p < 0.05) with a 57% of fibrous capsule formation inhibition. Thus, PRP inhibits fibrous capsule formation showing significant in vivo anti-angiogenic effects.
| 18 |
Anti-invasion, anti-migration and anti-EMT effect of PRP
To analyze the in vitro anti-metastatic effect of the proenzyme treatment, we studied the effect of PRP in cell invasion, cell migration and the modulation of EMT related genes in cancer cells. First, to evaluate the effect of PRP on cell migration, a key event in carcinogenesis, we performed a wound-healing assay on human pancreatic BxPC3 and human ovarian A2780 cells. Migration is defined as the directed movement of cells on a substrate such as plastic plates occurring on 2D surfaces.
Results show that non-treated cells migrated faster to close the gap of a scratch in the cell monolayer than PRP treated cells. PRP significantly reduced cell migration of pancreatic BxPC3 cells and compared with control cells even enhanced the width of the wound.

Although the A2780 ovarian tumor cell line does not grow forming a homogeneous monolayer like BxPC3, it can be observed that PRP treatment significantly reduces the ability of the ovarian cells to migrate. Data showed significant cell migration inhibition after 24 hours and 48 hours of treatment with PRP compared to control cells.
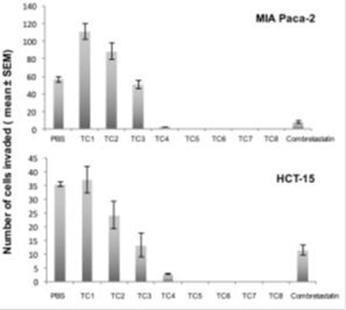
Secondly, we tested the inhibitory effect of the proenzyme formulation on cell invasion of colon and pancreatic tumor cells. Invasion is defined as cell movement through an extracellular 3D matrix. The principle of this assay is based on two medium containing chambers separated by a porous membrane through which cells transmigrate. Here, we tested different concentrations of PRP on MIA PaCa-2 pancreatic and HCT-15 colon human cancer cell lines. PRP showed a marked and significantly dose-dependent inhibition of invasion in both cell lines. Total inhibition of cell migration was achieved from PRP concentrations of T/C 0.015/0.093 mg/ml and so on with the other increasing concentrations tested.
| 19 |
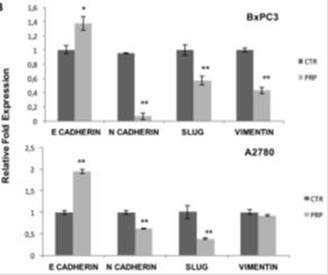
Finally, to investigate whether the exposure of PRP has a potential regulation in the transcriptional machinery that drives EMT in cancer cells, expression of EMT genes were studied in BxPC3 pancreatic and A2780 ovarian human cancer cells. EMT markers in both BxPC3 and A2780 cells were affected by PRP treatment at T/C 0.07/0.42 mg/ml. Results show that PRP treatment increased the expression of E-Cadherin (0.4 fold) (p < 0.05), whilst reduced the expressions of N-cadherin, Slug and vimentin (0.9, 0.5 and 0.6 fold, respectively) (p < 0.01) in BxPC3 cells.
In addition, PRP significantly up-regulated the expression of E-Cadherin (0.9 fold) (p < 0.01) and significantly down-regulated the expression of N-cadherin and Slug (0.4 and 0.6 fold, respectively) (p < 0.01) and induced a slight, but not significant, decrease of vimentin expression in A2780 cells.
PRP pharmacokinetic study
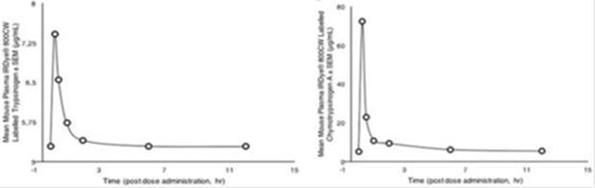
To evaluate the pharmacokinetics and organ distribution of trypsinogen and chymotrypsinogen, non-tumor bearing female athymic Nude-Foxn1nu mice were treated with IRDye® 800CW labeled trypsinogen (5 mg/kg) plus unlabeled trypsinogen (50 mg/kg), or IRDye® 800 CW labeled chymotrypsinogen (7 mg/kg) plus unlabeled Chymotrypsinogen (300 mg/kg). Animals were euthanized at specified time-points post-dose and plasma along with organ homogenates was prepared, then imaged via IVIS imaging system.
| 20 |
Fluorescence was measured in organ homogenates. Mice treated with labeled T, presented a fluorescence peak in all organs between 15 minutes and 2 hours post-dose. While mice treated with labeled C showed the maximum fluorescent emission between 15 minutes and 6 hours post-dose. For both highest readings were observed in the kidneys and liver. Maximum levels of both IRDye®800CW labeled trypsinogen and chymotrypsinogen A in mouse plasma occurred at 15 minutes post dose (7.5 and 72.2 ìg/ml, respectively). Levels of both IRDye® 800CW labeled proenzymes decreased rapidly after this time.
Anti-tumor efficacy of PRP in orthotropic mice models
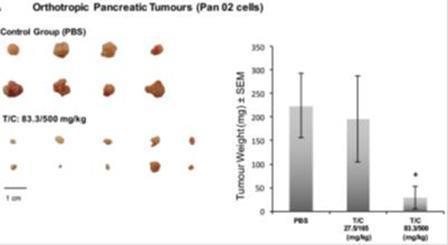
The effect of the proenzyme formulation PRP at different doses on tumor weight in orthotopically implanted pancreatic and ovary tumors was assessed. In the pancreatic tumor control group, there was significant (*P < 0.05) reduction in mean tumor weight in animals treated for 26 days with trypsinogen/chymotrypsinogen at 83.3/500 mg/kg (30.2 mg; 85.9% inhibition) compared with control (PBS; 214.8 mg), but not between trypsinogen/chymotrypsinogen at 27.5/165 mg/kg (196.5 mg; 8.5% inhibition) and the control (as shown in the figure below).
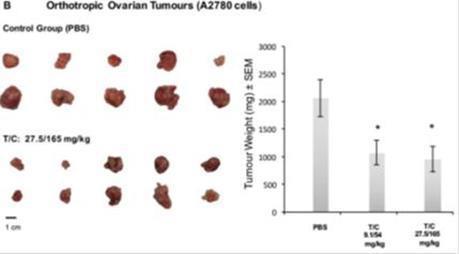
Furthermore, ovary tumor-bearing mice (as shown in the figure below) showed a significant (p < 0.05) reduction in mean tumor weight in animals treated for 21 days with two different doses of trypsinogen/chymotrypsinogen, 9.1/54 mg/kg and 27.5/165 mg/kg, compared with control (PBS). The mean weight of control group tumors was 2062.2 mg while the treated groups presented a mean tumor weight of 1074.2 mg and 957.3 respectively, ranging in a 50% tumor inhibition (52% - 46%).
| 21 |
The PRP Formulation
Oral pancreatic enzymes have been administered previously in a variety of circumstances and are in current clinical use in conditions where the pancreas is unable to produce sufficient enzymes for the digestion of food. A number of oral pancreatic enzyme products are presently approved in the U.S. for use in patients who do not produce enough pancreatic enzymes. Approved pancreatic enzyme products include Pancreaze™ from Johnson & Johnson, CREON® from Abbott Laboratories, and ULTRASE® from Axcan Pharma US.
PRP is a combination of two proenzymes, trypsinogen and chymotrypsinogen, specifically formulated within a specific ratio (1:6, as described above) designed to synergistically enhance their anti-cancer effects based on the mechanism of action. Patent protection for PRP has been secured in multiple jurisdictions, including the United States, and continues to be sought for similar compositions and mechanisms of action.
Oral enzymes have also been investigated previously for the treatment of cancer and, while generating encouraging results, their widespread use has been hampered by the very large quantities that have been considered necessary for effective treatment – 130 or more tablets per day. The high dose used with oral delivery is considered necessary due to the oral enzymes being broken down in the stomach and duodenum, the first part of the small intestine and very little actually being absorbed into the general circulation. By administering a proenzyme parenterally, and using a specific proenzyme formulation, the normal breakdown of the enzymes when taken orally is avoided and the drug can potentially be absorbed into the general circulation intact. It is also suggested that proenzymes are resistant to inactivation by numerous protein digesting enzymes, like serpins, which are circulating in the blood. Together with our scientific consultants, we believe that the development of a parenteral proenzyme formulation will lead to improved efficacy in the treatment of cancer compared with oral enzyme preparations, and will substantially reduce the dose in comparison to that used previously for oral enzyme therapy for the treatment of cancer.
Target Indications
The management of cancer differs widely, with a multitude of factors impacting the choice of treatment strategy. Some of those factors include:
| ● | the type of tumor, usually defined by the tissue in the body from which it originated; | |
| ● | the extent to which it has spread beyond its original location; |
| 22 |
| ● | the availability of treatments, driven by multiple factors including cost, drugs approved, local availability of suitable facilities, etc.; | |
| ● | regional and geographic differences; | |
| ● | whether the primary tumor is amenable to surgery, either as a potentially curative procedure, or as a palliative one; and | |
| ● | the balance between potential risks and potential benefits from the various treatments and, probably most importantly, the patient’s wishes. |
For many patients with solid cancers, such as breast, ovarian, colorectal, lung and pancreatic cancer, surgery is frequently the first treatment option, often followed by first line chemotherapy with or without radiotherapy. While hopefully such procedures are curative, in many instances the tumor returns, and second line treatment strategies are chosen in an effort to achieve a degree of control over the tumor. In most instances, the benefit is temporary, and eventually the point is reached where the patient’s tumor either fails to adequately respond to treatment, or the treatment has unacceptable toxicity which severely limits its usefulness.
Should the planned Phase I, II and III clinical trials confirm the efficacy of PRP, along with the favorable safety and tolerability profile suggested by pre-clinical studies conducted to date, we believe our product will have utility in a number of clinical situations including:
| ● | in the early stage management of solid tumors, most likely as part of a multi-pronged treatment strategy in combination with existing therapeutic interventions; | |
| ● | as a product that can be administered long term for patients following standard treatment approaches, such as surgery, or chemotherapy, in order to prevent or delay recurrence; and | |
| ● | as a preventative measure for patients at risk of developing cancer based on genetic screening. |
In the near term as part of our planned Phase I, II and III clinical trials, we plan to target patients with solid tumors, most likely ovarian and pancreatic, for whom other treatment options have been exhausted. This is a common approach by which most new drugs for cancer are initially tested. Once efficacy and safety has been demonstrated in this patient population, exploration of the potential utility of the drug in earlier stage disease can be undertaken, together with investigation of the drug’s utility in other types of cancers, such as gastro-esophageal tumors, colon or rectal carcinoma might be conducted. A Phase II study in a back-up indication, such as advanced therapy refractant prostate cancer will also be considered. This indication is based on positive preclinical pharmacology studies.
Development Strategy
Our goal is to undertake early stage clinical development of PRP through to a significant value inflection point, where the commercial attractiveness of a drug in development, together with a greater likelihood of achieving market authorization, may attract potential interest from licensees seeking to acquire new products. Such value inflection points in the context of cancer drugs are typically at the point where formal, controlled clinical trials have demonstrated either ‘efficacy’ or ‘proof of concept’ – typically meaning that there is controlled clinical trial evidence that the drug is effective in the proposed target patient population, has an acceptable safety profile, and is suitable for further development. From a ‘big picture’ perspective, it is our intention to progress the development of our technology through the completion of our planned Phase IIa clinical trials and then to seek a licensee for further development beyond that point.
| 23 |
As part of that commercial strategy, we will:
| ● | continue research and development to build our existing intellectual property portfolio, and to seek new, patentable discoveries; | |
| ● | seek to ensure all product development is undertaken in a manner that makes its products approvable in the major pharmaceutical markets, including the U.S., Europe, the UK, Australia and Japan; | |
| ● | aggressively pursue the protection of our technology through all means possible, including patents in all major jurisdictions, and potentially trade secrets; and | |
| ● | make strategic acquisitions to acquire new companies that have products or services that complement our future goals. |
Development Plan and Milestones
PRP
We plan to progress PRP down a conventional early stage clinical development pathway for:
| ● | regulatory and/or ethics approval to conduct a Phase Ib study, and submit it with the applicable government agency for approval; and | |
| ● | Phase IIa multiple escalating dose studies to investigate the safety, tolerability, and pharmacokinetics of PRP administered intravenously to patients. |
We are currently evaluating Australia, UK and Europe as the potential destination where we may commence the Phase Ib trial. In particular, we are closely evaluating Australia because of its research and development tax incentives, as well as a simplified regulatory environment. As part of such incentives, eligible companies conducting clinical trials in Australia receive up to 43.5% “cash-back” benefit in the form of a refund of their qualified research and development costs and expenses. The Company received a refund of $199,834 AUD ($134,728 USD) and $161,383 AUD ($115,437 USD) in the years ended June 30, 2020 and 2019 respectively. We are continuing to evaluate all options to conduct our planned clinical trials in the most cost-efficient manner, while striving to minimize dilution to our stockholders.
We anticipate reaching the Phase IIa proof of concept milestone in approximately three to four years, subject to regulatory approval in Europe, and the results from our research and development and licensing activities.
Our overhead and expenses are likely to increase from its current level as PRP progresses down the development pathway. This increase will be driven by the need to increase our internal resources in order to effectively manage our research and development activities.
| 24 |
Anticipated timelines
Below is the timeline and a detailed discussion of our anticipated milestones and steps that we plan to take in preparation for our planned Phase Ib clinical trial.
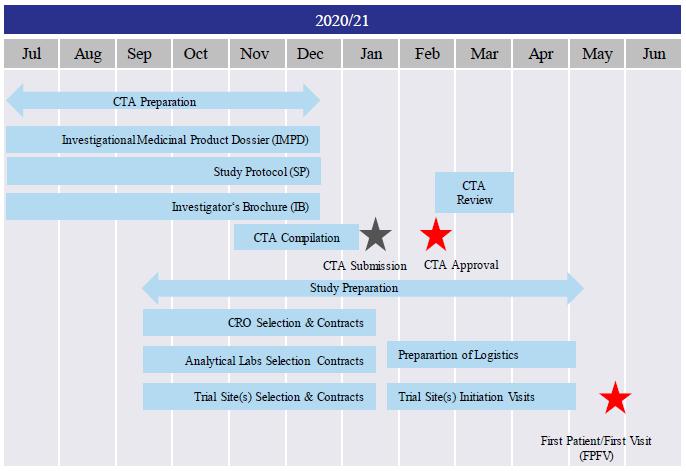
In first quarter of 2021 calendar year, we anticipate the submission of the Clinical Trial Application for PRP in a jurisdiction to be determined by us that would suit the best interests of the trial and our Company. We anticipate receiving approval of such application in the first calendar quarter of 2021. Following the clinical trial application, we plan to commence our Study Preparation, including CRO Selection and Contracts, Analytical Lab Selection Contracts and Trial Sites Selection and Contracts. In connection with the Clinical Trial Application, this product will be part of our Investigation Medicinal Product Dossier, Study Protocol and Investigator’s Brochure. In the first half of 2021 calendar year, we hope to complete the Study Preparation with the Preparation of Logistics and Trial Sites Initiation Visits and complete our clinical trial application review.
Commencing in the first half of 2021 calendar year, we intend to initiate a Phase Ib study in advanced cancer patients with solid tumors and the anticipated costs will be approximately $6.5 million. We will need to raise additional financing to fund our planned Phase I, II and III clinical trials and for working capital.
Financial Objectives
Multiple factors, many of which are outside of our control, can impact our ability to achieve our target objectives within the planned time and budgetary constraints. Subject to these caveats, our objective is to complete our planned Phase IIa study for PRP within the proposed budget.
Corporate Strategy
We primarily outsource services, skills and expertise to third parties as required to achieve our scientific and corporate objectives. As the business grows and gains more personnel, outsourcing will continue to be the preferred model, where fixed and variable costs are carefully managed on a project-by-project basis. This means our research and development activities are carried out by third parties. Additional third parties with specific expertise in research, compound screening and manufacturing (including raw material suppliers) have been contracted as required.
| 25 |
Our initial focus is to organize, coordinate and finance the various parts of our drug development pipeline. New personnel will be carefully introduced into our Company over a period of time as our research and development activities expand. They will have specific expertise in product development, manufacture and formulation, regulatory affairs, toxicology, clinical operations and business development (including intellectual property management, licensing and other corporate activities).
In the first instance, additional clinical management and development expertise is likely to be required for our lead product. Therefore, we anticipate an increase in employees in order to effectively manage our contractors as the projects progress down the development pathway.
This outsourcing strategy is common in the biotechnology sector, and is an efficient way to obtain access to the necessary skills required to progress a project, in particular as the required skills change as the project progresses from discovery, through manufacturing and non-clinical development and into clinical trials. We anticipate that we will continue to use this model, thereby retaining the flexibility to contract in the appropriate resource as and when required.
We intend to seek and identify potential licensing partners for our product candidates as they progress through the various development stages, reaching certain milestones and value inflection points. If a suitable licensee is identified, a potential licensing deal could consist of payments for certain milestones, plus royalties from future sales if the product is able to receive approval from the relevant regulatory authorities where future product sales are targeted. We intend to seek and identify potential licensees based on the initial efficacy data from Phase II clinical trials. To accomplish this objective, we have commenced discussions with potential partners in our current preclinical phase of development.
As part of our overall expansion strategy, from time to time, we investigate potential intellectual property acquisition opportunities to expand our product portfolio. While our initial focus is on the development of PRP as the lead product candidate, potential product candidates may also be considered for future preclinical and clinical development. These potential opportunities have arisen from other research and development organizations, which either own existing intellectual property or are currently developing new intellectual property, which may be of interest to us. These opportunities are possible new cancer treatments that are potentially less toxic than existing treatment approaches and are able to fill an existing gap in the treatment process, such as a systemic de-bulking method which could reduce the size and threat of metastases to a more manageable level for late stage cancer patients. We believe these potential treatment approaches will be complementary to existing treatment regimens and our existing product candidate, PRP. No formal approaches have been made at this stage and it is unknown whether we will engage in this discussion in the near future. However, we remain hopeful that as PRP progresses further down the development pathway, future opportunities may arise to use the expertise of our management and scientific personnel for future prospective research and development projects.
Current Operations
We are at a pre-revenue stage. We do not know when, if ever, we will be able to commercialize our products and begin generating revenue. We are focusing our efforts on organizing, coordinating and financing the various aspects of the drug research and development program outlined earlier in this document. In order to commercialize our products, we must complete preclinical development, Phase Ib, IIa and IIb clinical trials in Europe, the U.S., United Kingdom, Australia or elsewhere, and satisfy the applicable regulatory authority that PRP is safe and effective. If the results from the Phase II trials are convincing, we will seek conditional approval from the regulatory authorities sooner. Therefore, from the time we commence clinical trials, we estimate that this will take approximately three to four years if we seek conditional approval upon completion of Phase II trials, or up to seven years if we determine that Phase III trials are needed. As described previously, when we advance our development projects sufficiently down the development pathway and achieve a major increase in value, such as obtaining interim efficacy data from Phase II clinical trials, we will seek a suitable licensing partner to complete the remaining development activities, obtain regulatory approval and market the product.
| 26 |
Current Therapies
We are developing a therapeutic solution for the treatment of patients with advanced stages of cancer targeting solid tumors, which is cancer that originates in organs or tissues other than bone marrow or the lymph system. Common cancer types classified as solid tumors include lung, colorectal, ovarian cancer, pancreatic cancer and liver cancers. In each of these indications, there is a large market opportunity to capitalize on the limitations of current therapies.
Current therapeutic options for the treatment of cancer offer, at most, a few months of extra life or tumor stabilization. Some experts believe that drugs that kill most tumor cells do not affect cancer stem cells, which can regenerate the tumor (e.g. chemotherapy). Studies are revealing the genetic changes in cells that cause cancer and spur its growth. This research is providing scientific researchers with many potential targets for drugs. Tumor cells, however, can develop resistance to drugs.
Limitations of Current Therapies
PRP was developed because of the limitation of current cancer therapies. While surgery is often safe and effective for early stage cancer, many standard therapies for late stage cancer urgently need improvement; current treatments generally provide modest benefits, and frequently cause significant adverse effects. Our focus is to provide oncologists and their patients with therapies for metastatic cancer which are more effective than current therapies, and which have a substantially reduced side effect profile.
While progress has been made within the oncology sector in developing new treatments, the overall cancer death rate has only improved by 7% over the last 30 years. Most of these new treatments have some limitations, such as:
| ● | significant toxic effects; | |
| ● | expense; and | |
| ● | limited survival benefits. |
We believe that our treatment will provide a competitive advantage over the following treatments:
| ● | Chemotherapeutics: Side effects from chemotherapy can include pain, diarrhea, constipation, mouth sores, hair loss, nausea and vomiting, as well as blood-related side effects, which may include a low cell count of infection fighting white blood cells (neutropenia), low red blood cell count (anemia), and low platelet count (thrombocytopenia). Our goal is to demonstrate that our treatment will be more effective than chemotherapeutic and hormonal therapies with fewer side effects. | |
| ● | Targeted therapies: The most common type is multi-targeted kinase inhibitors (molecules which inhibit a specific class of enzymes called kinases). Common side effects include fatigue, rash, hand–foot reaction, diarrhea, hypertension and dyspnoea (shortness of breath). Furthermore, tyrosine kinases inhibited by these drugs appear to develop resistance to inhibitors. While the clinical findings with PRP are early and subject to confirmation in future clinical trials, no evidence has yet been observed of the development of resistance by the cancer to PRP. |
| 27 |
| ● | Monoclonal antibodies: Development of monoclonal antibodies is often difficult due to safety concerns. Side effects that are most common include skin and gastro-intestinal toxicities. For example, several serious side effects from Avastin, an anti-angiogenic cancer drug, include gastrointestinal perforation and dehiscence (e.g. rupture of the bowel), severe hypertension (often requiring emergency treatment) and nephrotic syndrome (protein leakage into the urine). Antibody therapy can be applied to various cancer types, but can also be limited to certain genetic sub populations in many instances. | |
| ● | Immunotherapy: There is a long history of attempts to develop therapeutic cancer vaccines to stimulate the body’s own immune system to attack cancer cells. While these products generally do not have the poor safety profile of standard therapeutic approaches, only a relatively small number of them are FDA-approved and available as compared to the number of patients diagnosed with cancer. Furthermore, only a relatively small number of the patient population is eligible to receive and subsequently respond to treatment, as defined by preventing tumor growth. |
License Agreements
We previously sponsored a collaborative research project at Bath University to investigate the cellular and molecular mechanisms underlying the potential clinical approach of our proprietary proenzyme formulation. As a result of this undertaking, we entered into a Commercialization Agreement with University of Bath (UK), dated November 12, 2009 (the “Commercialization Agreement”), where, initially, we held an exclusive license with Bath University, and where we and Bath University co-owned the intellectual property relating to our proenzyme formulations. The Commercialization Agreement originally provided for Bath University to assign the Patents (as defined therein) to Propanc in certain specified circumstances, such as successful completion of a clinical trial and commencement of a Phase II (Proof of Concept) clinical trial.
On June 14, 2012, Propanc and Bath University agreed to an earlier assignment to us of the patents pursuant to an Assignment and Amendment Deed, on the proviso that Bath University retains certain rights arising from the Commercialization Agreement, as follows:
| ● | Bath University reserves for itself (and its employees and students and permitted academic sub-licensees with respect to research use) the non-exclusive, irrevocable, worldwide, royalty free right to use the patents for research use; | |
| ● | The publication rights of Bath University specified in the contract relating to the original research made between the parties with an effective date of July 18, 2008 shall continue in force; | |
| ● | Propanc shall pay to Bath University a royalty of two percent of any and all net revenues; | |
| ● | Propanc shall use all reasonable endeavors to develop and commercially exploit the patents for the mutual benefit of Bath University and Propanc to the maximum extent throughout the covered territory and in any additional territory and to obtain, maintain and/or renew any licenses or authorizations that are necessary to enable such development and commercial exploitation. Without prejudice to the generality of the foregoing, Propanc shall comply with all relevant regulatory requirements in respect of its sponsoring and/or performing clinical trials in humans involving the administration of a product or materials within a claim of the patents; and |
| 28 |
| ● | Propanc shall take out with a reputable insurance company and maintain liability insurance coverage prior to the first human trials. |
In consideration of such assignment, we agreed to pay royalties of 2% of net revenues to Bath University. Additionally, we agreed to pay 5% of each and every license agreement subscribed for. The contract is cancellable at any time by either party. To date, no amounts are owed under the agreement.
We continue to learn the properties of proenzymes with the long-term aim of screening new compounds for development. We anticipate engaging in future discussions with several technology companies who are progressing new developments in the oncology field as potential additions to our product line. Initially targeting the oncology sector, our focus is to identify and develop novel treatments that are highly effective targeted therapies, with few side effects as a result of toxicity to healthy cells.
Intellectual Property
We have filed multiple patent applications relating to our lead product, PRP. The first application was filed in October 2010 in each of the countries listed in the table below. This application has been granted and remains in force in the United States, Belgium, Czech Republic, Denmark, France, Germany, Ireland, Italy, Netherlands, Portugal, Spain, Sweden, Switzerland, Liechtenstein, Turkey, United Kingdom, Australia, China, Japan, Indonesia, Israel, New Zealand, Singapore, Malaysia, South Africa, Mexico, Republic of Korea and India. In Brazil and Canada, the patent application remains under examination.
In 2016 and 2017 we filed other patent applications, as indicated below. Three applications were filed under the PCT. The PCT assists applicants in seeking patent protection by filing one international patent application under the PCT, which allows the applicants to seek protection for an invention in over 150 countries. Once national or regional applications are filed, the application is placed under the control of the national or regional patent offices, as applicable, in what is called the national or regional phase. One PCT application, filed in November 2016, entered the national phase in July 2018 in each of the countries listed in the table below. A second application filed in January 2017 entered the national phase commencing July 2018. A third application filed in April 2017 entered the national phase in October 2018.
| No. | Title | Country | Case Status | Date Filed | ||||
| 1. | A pharmaceutical composition for treating cancer comprising trypsinogen and/or chymotrypsinogen and an active agent selected from a selenium compound, a vanilloid compound and a cytoplasmic reduction agent. | USA, Belgium, Czech Republic, Denmark, France, Germany, Ireland, Italy, Netherlands, Portugal, Spain, Sweden, Switzerland, Liechtenstein, Turkey, United Kingdom, Australia, China, Japan, Indonesia, Israel, New Zealand, Malaysia, Singapore, Malaysia, South Africa, Mexico, Republic of Korea and India | Granted | Oct-22-2010 | ||||
| Brazil and Canada | Under Examination | |||||||
| USA | Two divisional applications granted and also under examination in Mexico and China | |||||||
| 2. | Proenzyme composition | Australia | Granted | Nov-11-2016 | ||||
| Canada, China, Europe, Hong Kong, India, Indonesia, Israel, Japan, Malaysia, New Zealand, Singapore, South Africa and USA | Application filed and pending | Nov-11-2016 | ||||||
| 3. | Cancer Treatment | Australia | Accepted | Jan-27-2017 | ||||
| Canada, China, Europe, Hong Kong, Israel, Japan, Malaysia, New Zealand, Singapore and USA | Application filed and pending | Jan-27-2017 | ||||||
| 4. | Composition of proenzymes for cancer treatment | Australia, China, Europe, Japan, and USA | Application filed and pending | Apr-12-2017 |
| 29 |
Further patent applications are expected to be filed to capture and protect additional patentable subject matter based on our field of technology relating to pharmaceutical compositions of proenzymes for treating cancer.
The basis of our intellectual property protection will be built around the following elements:
| ● | Method of use: Understanding the mechanism of action of the PRP proenzyme formulations, enabling the identification of new molecular targets, potential new therapeutic compounds and identification of new formulations that are adapted to enhance activity. | |
| ● | Formulation: We have developed an enhanced formulation containing the proenzyme trypsinogen in combination with at least one of two types of identified compounds considered effective for providing synergistic enhancement of the proenzyme formulations and different dosage ratios. A patentability assessment, based on an international prior art search, has indicated that strong potential exists for successfully obtaining patent claims covering the formulation. | |
| ● | Composition of Matter: Synthetic recombinant proteins designed to improve the quality, safety and performance of proenzymes used in the proposed formulations form part of the research and development program. |
Regulatory Matters
United States
Government oversight of the pharmaceutical industry is usually classified into pre-approval and post-approval categories. Most of the therapeutically significant innovative products marketed today are the subject of New Drug Applications (“NDA”). Preapproval activities, based on these detailed applications, are used to assure the product is safe and effective before marketing. In the United States, The Center for Drug Evaluation and Research (“CDER”), is the FDA organization responsible for over-the-counter and prescription drugs, including most biological therapeutics, and generic drugs.
Before approval, the FDA may inspect and audit the development facilities, planned production facilities, clinical trials, institutional review boards and laboratory facilities in which the product was tested in animals. After the product is approved and marketed, the FDA uses different mechanisms for assuring that firms adhere to the terms and conditions of approval described in the application and that the product is manufactured in a consistent and controlled manner. This is done by periodic unannounced inspections of production and quality control facilities by FDA’s field investigators and analysts.
Federal Food, Drug and Cosmetic Act and Public Health Service Act
Prescription drug and biologic products are subject to extensive pre- and post-market regulation by the FDA, including regulations that govern the testing, manufacturing, safety, efficacy, labelling, storage, record keeping, advertising and promotion of such products under the Federal Food, Drug and Cosmetic Act, the Public Health Service Act, and their implementing regulations. The process of obtaining FDA approval and achieving and maintaining compliance with applicable laws and regulations requires the expenditure of substantial time and financial resources. Failure to comply with applicable FDA or other requirements may result in refusal to approve pending applications, a clinical hold, warning letters, civil or criminal penalties, recall or seizure of products, partial or total suspension of production or withdrawal of the product from the market. FDA approval is required before any new drug or biologic, including a new use of a previously approved drug, can be marketed in the United States. All applications for FDA approval must contain, among other things, information relating to safety and efficacy, stability, manufacturing, processing, packaging, labelling and quality control.
New Drug Applications (“NDAs”)
The FDA’s NDA approval process generally involves:
| ● | Completion of preclinical laboratory and animal testing in compliance with the FDA’s good laboratory practice, or GLP, regulations; | |
| ● | Submission to the FDA of an investigational new drug (“IND”) application for human clinical testing, which must become effective before human clinical trials may begin in the United States; | |
| ● | Performance of adequate and well-controlled human clinical trials to establish the safety, purity and potency of the proposed product for each intended use; |
| 30 |
| ● | Satisfactory completion of an FDA pre-approval inspection of the facility or facilities at which the product is manufactured to assess compliance with the FDA’s “current good manufacturing practice” (“CGMP”) regulations; and | |
| ● | Submission to and approval by the FDA of a NDA. |
The preclinical and clinical testing and approval process requires substantial time, effort and financial resources, and we cannot guarantee that any approvals for our product candidates will be granted on a timely basis, if at all. Preclinical tests include laboratory evaluation of toxicity and immunogenicity in animals. The results of preclinical tests, together with manufacturing information and analytical data, are submitted as part of an IND application to the FDA. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions about the conduct of the clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin. Our submission of an IND may not result in FDA authorization to commence clinical trials. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development. Further, an independent institutional review board (“IRB”) covering each medical center proposing to conduct clinical trials must review and approve the plan for any clinical trial before it commences at that center and it must monitor the study until completed. The FDA, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive “good clinical practice” (“GCP”) regulations, which include requirements that all research subjects provide informed consent and that all clinical studies be conducted under the supervision of one or more qualified investigators.
For purposes of an NDA submission and approval, human clinical trials are typically conducted in the following sequential phases, which may overlap:
| ● | Phase I: Initially conducted in a limited population to test the product candidate for safety and dose tolerance; | |
| ● | Phase II: Generally conducted in a limited patient population to identify possible adverse effects and safety risks, to determine the initial efficacy of the product for specific targeted indications and to determine optimal dosage. A Phase IIa trial is a non-pivotal, exploratory study that assesses biological activity as its primary endpoint. A Phase IIb trial is designed as a definite dose finding study with efficacy as the primary endpoint. Multiple Phase II clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more extensive Phase III clinical trials; | |
| ● | Phase III: Commonly referred to as pivotal studies. When Phase II evaluations demonstrate that a dose range of the product is effective and has an acceptable safety profile, Phase III clinical trials are undertaken in large patient populations to further evaluate dosage, to provide substantial evidence of clinical efficacy and to further test for safety in an expanded and diverse patient population at multiple, geographically-dispersed clinical trial sites. Generally, replicate evidence of safety and effectiveness needs to be demonstrated in two adequate and well-controlled Phase III clinical trials of a product candidate for a specific indication. These studies are intended to establish the overall risk/benefit ratio of the product and provide adequate basis for product labelling; and |
| 31 |
| ● | Phase IV: In some cases, the FDA may condition approval of a NDA on the sponsor’s agreement to conduct additional clinical trials to further assess the product’s safety, purity and potency after NDA approval. Such post-approval trials are typically referred to as Phase IV clinical trials. |
Progress reports detailing the results of the clinical studies must be submitted at least annually to the FDA and safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events. Concurrent with clinical studies, sponsors usually complete additional animal studies and must also develop additional information about the product and finalize a process for manufacturing the product in commercial quantities in accordance with CGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Moreover, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
The results of product development, preclinical studies and clinical trials, along with the aforementioned manufacturing information, are submitted to the FDA as part of a NDA. NDA’s must also contain extensive manufacturing information. Under the Prescription Drug User Fee Act (“PDUFA”), the FDA agrees to specific goals for NDA review time through a two-tiered classification system, Standard Review and Priority Review. Standard Review is applied to products that offer at most, only minor improvement over existing marketed therapies. Standard Review NDAs have a goal of being completed within a ten-month timeframe, although a review can take significantly longer. A Priority Review designation is given to products that offer major advances in treatment, or provide a treatment where no adequate therapy exists. A Priority Review takes the FDA six months to review a NDA. It is likely that our product candidates will be granted Standard Reviews. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA may refer the application to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations.
The FDA may deny approval of a NDA if the applicable regulatory criteria are not satisfied, or it may require additional clinical data or additional pivotal Phase III clinical trials. Even if such data is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data from clinical trials is not always conclusive and the FDA may interpret data differently than Propanc. Once issued, product approval may be withdrawn by the FDA if ongoing regulatory requirements are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing, including Phase IV clinical trials, Risk Evaluation and Mitigation Strategies (“REMS”), and surveillance programs to monitor the effect of approved products that have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs. Products may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are any modifications to the drug, including changes in indications, labelling or manufacturing processes or facilities, approval of a new or supplemental NDA may be required, which may involve conducting additional preclinical studies and clinical trials.
Other U.S. Regulatory Requirements
After approval, products are subject to extensive continuing regulation by the FDA, which include company obligations to manufacture products in accordance with GMP, maintain and provide to the FDA updated safety and efficacy information, report adverse experiences with the product, keep certain records, submit periodic reports, obtain FDA approval of certain manufacturing or labeling changes and comply with FDA promotion and advertising requirements and restrictions. Failure to meet these obligations can result in various adverse consequences, both voluntary and FDA-imposed, including product recalls, withdrawal of approval, restrictions on marketing and the imposition of civil fines and criminal penalties. In addition, later discovery of previously unknown safety or efficacy issues may result in restrictions on the product, manufacturer or NDA holder.
Propanc, and any manufacturers of our products, are required to comply with applicable FDA manufacturing requirements contained in the FDA’s GMP regulations. GMP regulations require, among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation. The manufacturing facilities for our products must meet GMP requirements to the satisfaction of the FDA pursuant to a pre-approval inspection before Propanc can use them to manufacture products. Propanc and any third-party manufacturers are also subject to periodic inspections of facilities by the FDA and other authorities, including procedures and operations used in the testing and manufacture of our products to assess our compliance with applicable regulations.
| 32 |
With respect to post-market product advertising and promotion, the FDA imposes complex regulations on entities that advertise and promote pharmaceuticals, which include, among others, standards for direct-to-consumer advertising, promoting products for uses or in patient populations that are not described in the product’s approved labeling (known as “off-label use”), industry-sponsored scientific and educational activities and promotional activities involving the Internet. Failure to comply with FDA requirements can have negative consequences, including adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors and civil or criminal penalties. Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such off-label uses.
Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented. A NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing a NDA.
Adverse event reporting and submission of periodic reports is required following FDA approval of a NDA. The FDA also may require post-marketing testing, known as Phase IV testing, risk mitigation strategies and surveillance to monitor the effects of an approved product or to place conditions on an approval that could restrict the distribution or use of the product.
Orphan Drug Designation
In June 2017, we were notified by the FDA that PRP had been granted orphan drug designation for the treatment of pancreatic cancer. Orphan drug designation may be granted by the FDA when a rare disease or condition is implicated and a potential treatment qualifies under the Orphan Drug Act and applicable FDA regulations. This qualifies us for various developmental incentives, including protocol assistance, the potential for research grants, the waiver of future application fees, and tax credits for clinical testing if we choose to host future clinical trials in the United States.
In October 2017, we submitted a request for a second orphan drug designation for PRP, this time for ovarian cancer.
On November 2, 2017, we were notified by the FDA that our request was not granted. The Office of Orphan Products Development (“OOPD”) stated that complete prevalence is used as a measure of disease in ovarian cancer, as this reflects the number of women who have been diagnosed with disease and may be eligible for treatment with the proposed therapy. Therefore, on the date of the submission of our application, the OOPD estimated that the prevalence of ovarian cancer was 228,110 cases. Since the prevalence exceeds the threshold of 200,000 to qualify for orphan drug designation, they could not grant our request. We may consider resubmitting our application if we can identify a suitable sub population in ovarian cancer, which may meet the target threshold.
European Union
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials, commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or market our product in those countries. The approval process varies from country to country and the time may differ than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country. Despite these differences, the clinical trials will be conducted according to international standards such as Good Clinical Practice (GCP), Good Manufacturing Practice (GMP) and Good Laboratory Practice (GLP), which is recognized by each foreign country under the International Conference of Harmonization (ICH) Guidelines. We will conduct our trials in each foreign jurisdiction according to these standards, undertaking a First-In-Human (FIH) Phase I study in patients with advanced solid tumors, evaluating the safety, pharmacokinetics, and anti-tumor efficacy of PRP. This will be followed by two Phase II studies evaluating the efficacy and safety of PRP. To ensure harmonization between the jurisdictions, we intend to conduct regulatory meetings in the country where trials are conducted, as well as the FDA and European Medicines Agency. A pre-IND (Investigational New Drug) meeting will be held with the FDA once initial patient data has been collected from the FIH study to ensure acceptability of future planned Phase II trials.
| 33 |
Under European Union regulatory systems, we must submit and obtain authorization for a clinical trial application in each member state in which we intend to conduct a clinical trial. After we have completed clinical trials, we must obtain marketing authorization before it can market its product. We must submit applications for marketing authorizations for oncology products under a centralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. The European Medicines Agency (the “EMA”) is the agency responsible for the scientific evaluation of medicines that are to be assessed via the centralized procedure.
UK
On June 23, 2016, the UK government held a referendum to gauge voters’ support to remain or leave the European Union. The referendum resulted in 51.9% of UK voters in favor of leaving the European Union, commonly referred to as “Brexit.” On March 29, 2017, the UK invoked Article 50 of Lisbon Treaty to initiate complete withdrawal from the European Union, which was effected on January 31, 2020. The center for the EMA was based in London but the European Union has relocated the center to The Netherlands.
The impact of Brexit on the drug approval process in the UK is uncertain, which could significantly impact Propanc as we may elect to conduct our clinical trials for PRP in the UK. Companies based in the UK and operating in the drug industry are urging the European Union and the UK to reach an agreement to harmonize the regulatory process once the UK officially exits the European Union. We hope to commence our Phase IIa trials in 2021, and we are hopeful that there will be greater clarity on the regulatory process for drug approvals in UK in the near future.
Australia
In Australia, the relevant regulatory body responsible for the pharmaceutical industry is the Therapeutics Goods Administration (the “TGA”). Prescription medicines are regulated under the Therapeutic Goods Act 1989. Under the Therapeutic Goods Act, the Therapeutic Goods Administration evaluates new products for quality, safety and efficacy before being approved for market authorization, according to similar standards employed by the FDA and EMA in the United States and European Union, respectively. However, receiving market authorization in one or two regions does not guarantee approval in another.
Third-Party Payor Coverage and Reimbursement
Although none of our product candidates have been commercialized for any indication, if they are approved for marketing, commercial success of our product candidates will depend, in part, upon the availability of coverage and reimbursement from third-party payors at the federal, state and private levels. In addition, in many countries outside the United States, a drug must be approved for reimbursement before it can be approved for sale in that country.
Eligibility for reimbursement does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover costs and may not be made permanent. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies.
| 34 |
In many countries outside the United States, a drug must be approved for reimbursement before it can be approved for sale in that country. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. We may not obtain foreign regulatory approvals on a timely basis, if at all. We may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any foreign market.
Marketing Approvals, Pricing and Reimbursement Regulations
The regulations that govern marketing approvals, pricing and reimbursement for new drug products vary widely from country to country. In the United States, recently passed legislation may significantly change the approval requirements in ways that could involve additional costs and cause delays in obtaining approvals. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted.
Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels. A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products.
Other Regulations
We are also subject to numerous federal, state and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control, and disposal of hazardous or potentially hazardous substances. We may incur significant costs to comply with such laws and regulations now or in the future.
Competition
The biotechnology and pharmaceutical industries are characterized by continuing technological advancement and significant competition. While we believe that our technology platforms, product candidates, know-how, experience and scientific resources provide us with competitive advantages, we face competition from major pharmaceutical and biotechnology companies, academic institutions, governmental agencies and public and private research institutions, among others. Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future. Key product features that would affect our ability to effectively compete with other therapeutics include the efficacy, safety and convenience of our products. The level of generic competition and the availability of reimbursement from government and other third-party payers will also significantly impact the pricing and competitiveness of our products. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
| 35 |
Employees
As of October 6, 2020, we have one full-time employee and two part-time employees. In addition to our employees, we engage key consultants and utilize the services of independent contractors to perform various services on our behalf. Some of our executive officers and directors are engaged in outside business activities that we do not believe conflict with our business. Over time, we may be required to hire additional employees or engage independent contractors to execute various projects that are necessary to grow and develop our business. These decisions will be made by our officers and directors, if and when appropriate.
Our Corporate Information
Our principal executive office is located at 302, 6 Butler Street, Camberwell, VIC, 3124 Australia.
Available Information
Copies of our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and other documents that we will file with or furnish to the SEC will be available free of charge by sending a written request to our Corporate Secretary at our corporate headquarters. Additionally, the documents we file with the SEC are or will be available free of charge at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549. Other information on the operation of the Public Reference Room may be obtained by calling the SEC at (800) SEC-0330. The SEC maintains a website that contains reports, proxy and information statements and other information regarding registrants that file electronically with the SEC. The SEC’s website is www.sec.gov.
We maintain a corporate website at www.propanc.com. You will be able to access our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and amendments to those reports, proxy statements and other information to be filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act with the SEC free of charge at our website as soon as reasonably practicable after such material will be electronically filed with, or furnished to, the SEC. The information contained in, or that can be accessed through, our website is not part of this Annual Report.
Our telephone number is +61-03-9882-6723 and our website is www.propanc.com. Unless expressly noted, none of the information on our website is part of this prospectus or any prospectus supplement. Our common stock is quoted on the OTCQB market operated by the OTC Markets Group, Inc., or “OTCQB,” under the ticker symbol “PPCB.”
Summary of Risks
Our business is subject to a number of risks and uncertainties that you should understand before making an investment decision. For example, we have no commercial product, a history of net losses, we expect to continue to incur net losses, we will require significant additional funding and we may not achieve or maintain profitability. Furthermore, we have no cash flow from operations to sustain our operations. We have historically relied upon the issuance of equity and/or convertible debt to fund our operations, which debt we are currently unable to repay in cash. Our ability to ever generate revenues will depend solely on the commercial success of PRP, our only prospective product, which depends upon its approval by applicable regulatory authorities and then market acceptance by purchasers in the pharmaceutical market and the future market demand and medical need for products and research utilizing PRP. At present, PRP has only been used for research and clinical trial purposes in animals, and there is no commercially approved drug product or drug product submitted in a pending marketing application that incorporates PRP as an ingredient. As a result, no marketing authority has reviewed our drug master file (DMF) for PRP as a product ingredient or inspected our Company. As of June 30, 2020, we have an accumulated deficit of approximately $56 million since inception. We have incurred substantial net losses since our inception, including net loss of $4,740,723 and $5,758,369 for the fiscal years ended June 30, 2020 and June 30, 2019, respectively. We expect to incur additional losses as we continue to invest in our research and development programs and move forward with our human clinical trials application, clinical trials and commercialization activities. Additional risks are discussed more fully in the section entitled “Risk Factors” following this prospectus summary. These risks include, but are not limited to, the following:
| ● | Our ability to continue as a going concern absent obtaining adequate new debt and/or equity financings. |
| 36 |
| ● | We face risks related to Novel Coronavirus (COVID-19) which could significantly disrupt our research and development, operations, sales, and financial results. | |
| ● | We have incurred significant losses since our inception, and we expect to incur significant losses for the foreseeable future and may never achieve or maintain profitability. | |
| ● | We will continue to need substantial additional funding and raise capital when needed to initiate and continue our product development programs and commercialization efforts. | |
| ● | As an early stage company, it may be difficult for you to evaluate the success of our business to date and to assess our future viability. | |
| ● | We currently rely, and may continue to rely for the foreseeable future, on substantial debt financing that we are not able to repay in cash. | |
| ● | Raising additional capital is highly likely to cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidate. | |
| ● | The conversion of some or all of our currently outstanding convertible notes in shares of our common stock will dilute the ownership interests of existing stockholders. | |
| ● | It may be difficult for you to evaluate the success of our business to date and to assess our future viability. | |
| ● | Our only product candidate, PRP, remains in the early stages of development and may never become commercially viable, and therefore, you may lose your investment. | |
| ● | PRP may cause undesirable side effects that could negatively impact its clinical trial results or limit its use, hindering further development, subject us to possible product liability claims, and make it more difficult to commercialize PRP. | |
| ● | Our ability to successfully initiate and complete our clinical trials of PRP. | |
| ● | Our ability to obtain regulatory approval in jurisdictions in the United States and outside the United States to be able to market PRP in those jurisdictions. | |
| ● | Our ability in the future to establish sales and marketing capabilities or enter into agreements with third parties to sell and market PRP. | |
| ● | We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do. | |
| ● | Our ability to seek approval for reimbursement for PRP before it can be marketed, assuming successful commercialization, and us being then subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives. | |
| ● | We may depend on collaborations with third parties for the development and commercialization of PRP, and these collaborations may be unsuccessful. | |
| ● | Our third party manufacturers of PRP performing satisfactorily or at all, and our reliance on any third-party for the supply of PRP. | |
| ● | Our ability to comply with our obligations under any intellectual property licenses with third parties. | |
| ● | Our ability to protect our intellectual property rights. | |
| ● | Our ability to obtain, or if there are delays in obtaining, required regulatory approvals, to commercialize PRP, and our ability to generate revenue. | |
| ● | Our ability to obtain marketing approval in international jurisdictions to market PRP in international jurisdictions. | |
| ● | Our ability to obtain marketing approval of and commercialize PRP and affect the prices we may obtain. |
| 37 |
| ● | PRP or any other product candidate for which we obtain marketing approval could be subject to restrictions or withdrawal from the market and our ability to comply with applicable regulatory requirements. | |
| ● | We rely on the significant experience and specialized expertise of the Chief Executive Officer and the Chief Financial Officer who works on a part-time bases, we do not currently have any other members of a management team. | |
| ● | We have identified material weaknesses in our internal control over financial reporting that, if not properly remediated, could result in material misstatements in our consolidated financial statements in future periods. | |
| ● | We do not have any independent directors, which represents a potential conflict of interest, and helps create a material weakness in our disclosure controls and procedures as well as our internal control over financial reporting. | |
| ● | Our ability to implement and maintain an effective system of internal control over financial reporting, and accordingly, our ability to accurately report our financial results or prevent fraud. | |
| ● | The market price of our common stock may continue to be highly volatile, and you may not be able to resell your shares at or above the public offering price and therefore, you could lose all or part of your investment. | |
| ● | Our shares of common stock are thinly traded and there may not be an active, liquid trading market for our common shares. | |
| ● | Our Chief Executive Officer is our controlling shareholder and will continue to control our Company for the foreseeable future due to his ownership of super-voting shares, and therefore, it is not likely that you will be able to elect directors or have any say in the policies of our Company. | |
| ● | Future sales and issuances of our capital stock or rights to purchase capital stock will result in additional dilution of the percentage ownership of our stockholders and could cause our stock price to decline. | |
| ● | We are a smaller reporting company, and therefore, we are subject to scaled disclosure requirements that may make it more challenging for investors to analyze our results of operations and financial prospects. |
| 38 |
THE OFFERING
| Common Stock registered in this offering: | The Selling Shareholder is offering up to 150,000,000 shares of Common Stock, including shares of Common Stock underlying the Warrants that may held by the Selling Stockholder, consisting of (i) 1.5 shares of the Company’s common stock (the “Common Stock”), or pre-funded warrants (the “Pre-funded Warrants”) and (ii) 1.5 warrants to purchase one share of Common Stock (“Series A Warrants”, and collectively with the Common Stock the “Units”). Each Series A Warrant has an exercise price per share equal to $0.20 per share. The Series A Warrants will be exercisable immediately and will expire on the three-year anniversary of its original issuance date. The purchaser is not permitted to exercise portion of the Series A Warrant that would result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% of our outstanding common stock following the consummation of this offering.
The purchaser is not permitted to exercise portion of the Pre-funded Warrant that would result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% of our outstanding common stock following the consummation of this offering. Each pre-funded warrant included in the pre-funded units will have a per share exercise price of $0.0001. The pre-funded warrants contained in the pre-funded units will be exercisable immediately and may be exercised at any time until the Pre-funded Warrants are exercised in full. This offering also relates to the shares of common stock issuable upon exercise of any Pre-funded Warrants contained in the Units sold in this offering.
Because we will issue either Series A Warrants or Pre-Funded Warrants as part of each Unit, the number of warrants sold in this offering will not change as a result of a change in mix of the Series A Warrants and Pre-Funded Warrants sold. |
| Common warrants registered in the offering | The Selling Stockholder is offering to 63,750,000 of each of the following common stock purchase warrants; Series B Warrants and Series C Warrants. Each Series B Warrant has an exercise price per share equal to $0.04 per share and will expire on the three-year anniversary of its original issuance date. The Purchaser is not permitted to exercise portion of the Series B Warrants that would result in the Purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% of our outstanding common stock following the consummation of this offering.
Each Series C Warrant has an exercise price per share equal to $0.20 per share. The purchaser is not permitted to exercise portion of the Series C Warrants that would result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% of our outstanding common stock following the consummation of this offering. |
Selling Stockholder |
Ionic Ventures, LLC |
| Use of proceeds | We will not receive any proceeds from the sale of common stock by the Selling Stockholder. All of the net proceeds from the sale of our common stock will go to the Selling Stockholder as described below in the sections entitled “Selling Stockholder” and “Plan of Distribution”. See “Use of Proceeds” on 66 of this prospectus. |
| Risk factors | Investing in our securities is highly speculative and involves a high degree of risk. You should carefully consider the information set forth in the “Risk Factors” section beginning on page 40 before deciding to invest in our securities. |
| Trading symbol | Our common stock is currently quoted on the OTCQB under the trading symbol “PPCB”. |
| 39 |
Investing in our common stock involves a high degree of risk. You should carefully consider the risks described below, as well as the other information in this prospectus, including our consolidated financial statements and the related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” before deciding whether to invest in our common stock. The occurrence of any of the events or developments described below could harm our business, financial condition, operating results, and growth prospects. In such an event, the market price of our common stock could decline, and you may lose all or part of your investment. Additional risks and uncertainties not presently known to us or that we currently deem immaterial also may impair our business operations.
RISKS RELATED TO THIS OFFERING AND OWNERSHIP OF OUR SECURITIES
An investment in shares of common stock, the units and the warrants are extremely speculative and there can be no assurance of any return on any such investment.
An investment in the shares of common stock, units and the warrants is extremely speculative and there is no assurance that investors will obtain any return on their investment. Investors will be subject to substantial risks involved in an investment in us, including the risk of losing their entire investment.
Holders of our warrants will have no rights as a common stockholder until they acquire our shares of common stock.
Until you acquire our shares of common stock upon exercise of your warrants, you will have no rights with respect to our shares of common stock issuable upon exercise of your warrants. Upon exercise of your warrants, you will be entitled to exercise the rights of a common stockholder only as to matters for which the record date occurs after the exercise date.
The warrants issued in this offering may not have any value.
The 11,250,000 Series A Warrants each have an exercise price equal to $0.20 and will expire on the three (3) year anniversary of the date they first become exercisable. The 63,750,000 Series B Warrants each have an exercise price equal to $0.04 and will expire on the three (3) year anniversary of the date they first become exercisable. The 63,750,000 Series C Warrants each have an exercise price equal to $0.20 and will expire on the three (3) year anniversary of the date they first become exercisable.
| 40 |
If our shares of common stock are not listed on a national securities exchange, U.S. holders of the warrants may not be able to exercise their warrants without compliance with applicable state securities laws and the value of your warrants may be significantly reduced.
Our shares of common stock are currently quoted on the OTCQB. If our shares of common stock are not listed on a national securities exchange at the time of the completion of this offering, the exercise of the warrants by U.S. holders may not be exempt from state securities laws. As a result, depending on the state of residence of a holder of the warrants, a U.S. holder may not be able to exercise its warrants unless we comply with any state securities law requirements necessary to permit such exercise or an exemption applies. Although we plan to use our reasonable efforts to assure that U.S. holders will be able to exercise their warrants under applicable state securities laws if no exemption exists, there is no assurance that we will be able to do so. As a result, in the event that our shares of common stock are not listed on a national securities exchange at the time of the completion of this offering, your ability to exercise your warrants may be limited. The value of the warrants may be significantly reduced if U.S. holders are not able to exercise their warrants under applicable state securities laws.
We will require additional financing or financings, which would result in substantial dilution to existing stockholders.
Without additional financing or curtailing our operations, we may not have the operating capital to continue our operations through 2021. Management expects to continue incurring losses for the foreseeable future and will need to raise additional capital to pursue our business plan into 2020. This offering is part of such efforts to raise additional required capital. In addition, we may decide to expand operations, undertake strategic acquisitions or determine some other business need. Financing could include debt and/or equity financings, including transactions with strategic customers and partners that may include debt and/or equity arrangements. Such sources of financing may not be available on acceptable terms, if at all. Failure to obtain such financing may cause us to curtail or cease operations and/or result in delay or indefinite postponement of research, development and launch of PRP, expansion initiatives, capital expenditures and other operational priorities. Any transaction involving the issuance of previously authorized but unissued shares of common stock, or securities convertible into shares of common stock, are likely to result in dilution, possibly substantial, to present and prospective holders of shares of common stock and may be on unfavorable to us.
We face risks related to Novel Coronavirus (COVID-19) which could significantly disrupt our research and development, operations, sales, and financial results.
Our business will be adversely impacted by the effects of the Novel Coronavirus (COVID-19). In addition to global macroeconomic effects, the Novel Coronavirus (COVID-19) outbreak and any other related adverse public health developments will cause disruption to our operations, research and development, and sales activities. Our third-party manufacturers, third-party distributors, and our customers have been and will be disrupted by worker absenteeism, quarantines and restrictions on employees’ ability to work, office and factory closures, disruptions to ports and other shipping infrastructure, border closures, or other travel or health-related restrictions. Depending on the magnitude of such effects on our activities or the operations of our third-party manufacturers and third-party distributors, the supply of our products will be delayed, which could adversely affect our business, operations and customer relationships. In addition, the Novel Coronavirus (COVID-19) or other disease outbreak will in the short-run and may over the longer term adversely affect the economies and financial markets of many countries, resulting in an economic downturn that will affect demand for our products and impact our operating results. There can be no assurance that any decrease in sales resulting from the Novel Coronavirus (COVID-19) will be offset by increased sales in subsequent periods. Although the magnitude of the impact of the Novel Coronavirus (COVID-19) outbreak on our business and operations remains uncertain, the continued spread of the Novel Coronavirus (COVID-19) or the occurrence of other epidemics and the imposition of related public health measures and travel and business restrictions will adversely impact our business, financial condition, operating results and cash flows. In addition, we have experienced and will experience disruptions to our business operations resulting from quarantines, self-isolations, or other movement and restrictions on the ability of our employees to perform their jobs that may impact our ability to develop and design our products in a timely manner or meet required milestones or customer commitments.
| 41 |
RISKS RELATED TO OUR FINANCIAL CONDITION AND OUR NEED FOR ADDITIONAL CAPITAL
Our ability to continue as a going concern is in substantial doubt absent obtaining adequate new debt or equity financings.
We have concerns about our ability to continue as a going concern based on the absence of revenues, recurring losses from operations and our need for additional financing to fund all of our operations. Working capital limitations continue to impinge on our day-to-day operations, thus contributing to continued operating losses. For the fiscal years ended June 30, 2020 and June 30, 2019, we had net losses of $4,740,723 and $5,758,369, respectively. Further, as of June 30, 2020, we had $67,007 in cash and had an accumulated deficit of $55,781,770.
Based upon our current business plan, we will need considerable cash investments to have the opportunity to be successful. Our capital requirements and cash needs are significant and continuing. We can provide no assurance that we will be able to generate a sufficient amount of revenue, if any, from our business in order to achieve profitability. It is not possible at this time for us to predict with assurance the potential success of our business. The revenue and income potential of our proposed business and operations are unknown. If we cannot continue as a viable entity, we may be unable to continue our operations and you may lose some or all of your investment in our common stock.
We have incurred significant losses since our inception. We expect to incur significant losses for the foreseeable future and may never achieve or maintain profitability.
Since inception, we have incurred significant operating losses. Our net loss was $4,740,723 and $5,758,369, respectively, for the fiscal years ended June 30, 2020 and June 30, 2019 respectively. As of June 30, 2020, we had a deficit accumulated of $55,781,770. To date, we have not generated any revenues and have financed most of our operations with funds obtained from private financings.
Since October 2007, we have devoted substantially all of our efforts to research and development of our product candidates, particularly PRP, and efforts to protect our intellectual property. Most recently, from January-February 2016, and October 2016-April 2017, we have contracted with third parties to perform a number of laboratory studies and dose range finding studies designed to examine the anti-cancer effects of PRP and prepare for human clinical trials. Since mid-2017, we developed a suitable manufacturing process for each active drug substance in the PRP formulation, capable of producing a full scale GMP manufacture of PRP for human trials. We were granted Orphan Drug Designation status from the FDA for PRP for the treatment of pancreatic cancer. In March 2018, a scientific advice meeting was conducted with the MHRA (Medicines and Healthcare Products Regulatory Agency) UK, to assist with preparation of our first Clinical Trial Application (CTA). We expect that it will be many years, if ever, before we have a product candidate ready for commercialization. We expect to incur significant expenses and increasing operating losses for the foreseeable future if and as we progress PRP into clinical trials, continue our research and development, seek regulatory approvals, establish or contract for a sales and marketing infrastructure, maintain and expand our intellectual property portfolio, and add personnel.
To become profitable, we must develop and eventually commercialize PRP or some other product with significant market potential. This will require us to successfully complete clinical trials, obtain market approval and market and sell PRP or whatever other product that we obtain approval for. We might not succeed in any one or a number of these activities, and even if we do, we may never generate revenues that are significant enough to achieve profitability. Our failure to become and remain profitable would decrease our value and could impair our ability to raise capital, maintain our research and development efforts, expand our business or continue our operations.
| 42 |
As an early stage company, it may be difficult for you to evaluate the success of our business to date and to assess our future viability.
Despite having been founded in 2007, we remain an early-stage company. We commenced active operations in the second half of 2010. Our operations to date have been mainly limited to establishing our research programs, particularly PRP, building our intellectual property portfolio and deepening our scientific understanding of our product development. We have not yet initiated, let alone demonstrated any ability to successfully complete, any clinical trials, including large-scale, pivotal clinical trials, obtain marketing approvals, manufacture a commercial scale product, or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization. It will take a number of years for PRP to be made available for the treatment of cancer, if it ever is. Given our relatively short operating history compared to the timeline required to fully develop a new drug, you are cautioned about making any predictions on our future success or viability based on our activities or results to date. In addition, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown factors. We will eventually need to transition from a company with a research focus to a company capable of supporting commercial activities. We may not be successful in such a transition.
We currently rely, and may continue to rely for the foreseeable future, on substantial debt financing that we are not able to repay in cash.
In order to maintain our operations, including our research and development efforts and our preclinical development of PRP, we have over the last few years entered into a number of securities purchase agreements pursuant to which we issued convertible debt in return for cash. We are not currently able to repay either the current principal or interest on this debt in cash. Our lenders, therefore, can convert their debt into shares of our common stock, at a discount to current market prices and then attempt to sell these shares on the open market in order to pay down their loans and receive a return on their investment. These financings pose the risk that as these debts are converted, our stock price will reflect the reduced prices our lenders are willing to sell their shares at, given the discount they have received. These financings contain no floor on the price our lenders can convert their debt into shares of our common stock and they could conceivably reduce the price our common stock to near zero. These types of financings negatively impact our balance sheet and the appeal of our common stock as an investment. While we are actively exploring various alternatives to reduce if not eliminate this debt, for the foreseeable future we will continue to carry it on our balance sheet, and we may have to enter into additional such financings in order to sustain our operations. As a result, the price of our common stock and our market capitalization are subject to significant declines until our convertible debt is either refinanced on a favorable basis or is eliminated.
As of June 30, 2020, the total amount of debt outstanding under these convertible notes, including interest, is approximately $1,100,000 (not including redemption premium). Please see the section captioned “Management’s Discussion of Financial Condition and Results of Operations - Recent Developments” for further information
We will continue to need substantial additional funding. If we are unable to raise capital when needed, we would be forced to delay, reduce or eliminate our product development programs or commercialization efforts.
We expect our expenses to significantly increase in connection with our ongoing activities, particularly if we initiate clinical trials of, and ultimately seek marketing approval for, PRP. In addition, even if we ultimately obtain marketing approval for PRP or any other product candidate, we expect to incur significant commercialization expenses related to product sales, marketing, manufacturing and distribution. We also hope to continue and expand our research and development activities. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce or eliminate our future commercialization efforts or any research and development programs.
Our future capital requirements will depend on many factors, including, among others, the scope, progress and, results of our potential future clinical trials, the costs, timing and outcome of regulatory review of PRP, the costs of any future commercialization activities, and the costs of preparing and filing future patent applications, if any. Accordingly, we will continue to rely on additional financing to achieve our business objectives. Adequate additional financing may not be available to us on acceptable terms, or at all. Even if we are able to enter into financing agreements, we may be forced to pay higher interest rates, accept default provisions in financing agreements that we believe are overly punitive, make balloon payments as required, and, as noted below, if we issue convertible debt the price of our common stock may well be negatively affected and our existing stockholders may suffer dilution.
| 43 |
Raising additional capital will cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
Until such time, if ever, as we can generate substantial product revenues, we expect to continue to finance our cash needs through a combination of equity offerings and additional debt financings, and possibly also through future collaborations, strategic alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or debt securities, including convertible debt securities, the ownership interest of our existing stockholders will be diluted upon conversion, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of our existing stockholders.
Debt financing, if available, may also involve agreements that include restrictive covenants limiting or restricting our ability to take specific actions, such as merging with other companies or consummating certain changes of control, acquiring other companies, engaging in new lines of business, incurring additional debt, making capital expenditures, making certain investments, paying dividends, transferring or disposing of assets, amending certain material agreements, incurring additional indebtedness or enter into various specified transactions. We therefore may not be able to engage in any of the foregoing transactions unless we obtain the consent of the lender or terminate such debt agreements. Our debt agreements may also contain certain financial covenants, including achieving certain milestones and may be secured by substantially all of our assets. In the event we enter into such debt agreements, there is no guarantee that we will be able to generate sufficient cash flow or sales to pay the principal and interest under our debt agreements or to satisfy all of the financial covenants.
If we raise additional funds through collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
The conversion of some or all of our currently outstanding convertible notes in shares of our common stock will dilute the ownership interests of existing stockholders.
The conversion of some or all of our currently outstanding convertible notes in shares of our common stock will dilute the ownership interests of existing stockholders. As of June 30, 2020, we had 11 outstanding notes convertible into approximately 439,113,000 shares of our common stock (based on then applicable conversion prices). Each holder of the notes has agreed to a 4.99% beneficial ownership conversion limitation (subject to certain noteholders’ ability to increase such limitation to 9.99% upon 60 days’ notice to us), and each note may not be converted during the first six-month period from the date of issuance. Any sales in the public market of the common stock issuable upon such conversion or any anticipated conversion of our convertible notes into shares of our common stock could adversely affect prevailing market prices of our common stock.
The accounting method for convertible debt securities that may be settled in cash could have a material adverse effect on our reported financial results.
Under Financial Accounting Standards Board Accounting Standards Codification 470-20, Debt with Conversion and Other Options (“ASC 470-20”), we are required to separately account for the liability and equity components of our convertible notes because they may be settled entirely or partially in cash upon conversion in a manner that reflects our economic interest cost. The effect of ASC 470-20 on the accounting for our convertible notes is that the equity component is required to be included in the additional paid-in capital section of stockholders’ deficit on our consolidated balance sheet, and the value of the equity component would be treated as a discount for purposes of accounting for the debt component of our convertible notes. As a result, we will be required to record a greater amount of non-cash interest expense in current periods presented as a result of the amortization of the discounted carrying value of our convertible debt or notes to their face amount over the terms. We will report higher net loss in our financial results in part because ASC 470-20 will require interest to include both the current period’s amortization of the debt discount and the instrument’s coupon interest, which could adversely affect our reported or future financial results, the trading price of our common stock and the trading price of our convertible notes.
| 44 |
In addition, because our convertible notes may be settled entirely or partly in cash, under certain circumstances, these are currently accounted for utilizing the treasury stock method, the effect of which is that the shares issuable upon conversion are not included in the calculation of diluted earnings per share except to the extent that the conversion value exceeds their principal amount. Under the treasury stock method, for diluted earnings per share purposes, the transaction is accounted for as if the number of shares of common stock that would be necessary to settle such excess, if we elected to settle such excess in shares, are issued. We cannot be sure that the accounting standards in the future will continue to permit the use of the treasury stock method. If we are unable to use the treasury stock method in accounting for the shares issuable upon conversion of our convertible notes, then our diluted earnings per share would be adversely affected.
We maintain our cash in Australian financial institutions that are not insured.
The Company maintains its cash in banks and financial institutions in Australia. Bank deposits in Australian banks are uninsured. The Company has not experienced any losses in such accounts through to date.
RISKS RELATED TO THE DISCOVERY, DEVELOPMENT AND COMMERCIALIZATION OF OUR PRODUCT CANDIDATES
Because PRP remains in the early stages of development and may never become commercially viable, you may lose your investment.
At present, our only product candidate, PRP, is still in preclinical development. While we are hopeful that the preclinical testing we have completed will lead to our initiating human clinical trials in 2020, as noted elsewhere we expect that it will be several years, at least, before PRP can be commercialized. Further, if clinical trials for PRP fail to produce statistically significant results, we would likely be forced to either spend several more years in development attempting to correct whatever flaws were identified in the trials, or we would have to abandon PRP altogether. Either of those contingencies, and especially the latter, would dramatically increase the amount of time before we would be able to generate any product-related revenue, and we may well be forced to cease operations. Under such circumstances, you may lose at least a portion of, and perhaps your entire, investment.
PRP may cause undesirable side effects that could negatively impact its clinical trial results or limit its use, hindering further development, subject us to possible product liability claims, and make it more difficult to commercialize PRP.
In addition to the possibility that the clinical trials we hope to initiate for PRP could demonstrate a lack of efficacy, if we alternatively identify adverse and undesirable side effects caused by it this will likely interrupt, delay or even halt our further development, or possibly limit our planned therapeutic uses for it, and may even result in adverse regulatory action by the FDA or other regulatory authorities.
Moreover, this may subject us to product liability claims by the individuals enrolled in our clinical trials; while we intend to obtain product liability insurance in connection with our clinical trials, it is possible that the potential liability of any claims against us could exceed the maximum amount of this coverage, or at least increase our premiums. Either would result in an increase in our operating expenses, in turn making it more difficult to complete our clinical development, or in the suspension or termination of the clinical trial. Any negative information concerning PRP, however unrelated to its composition or method of use, could also damage our chances to obtain regulatory approval.
Even if we are able to complete PRP’s development and receive regulatory approvals, undesirable side effects could prevent us from achieving or maintaining market acceptance of the product or substantially increase the costs and expenses of commercializing it.
Because successful development of our products is uncertain, our results of operations may be materially harmed.
Our development of PRP and future product candidates is subject to the risks of failure inherent in the development of new pharmaceutical products that are based on new technologies, including but not limited to delays in product development, clinical testing or manufacturing; unplanned and higher expenditures; adverse findings relating to safety or efficacy; failure to receive regulatory approvals; the emergence of superior or equivalent products; an inability by us or one of our collaborators to manufacture our product candidates on a commercial scale on our own, or in collaboration with third parties; and, ultimately, a failure to achieve market acceptance.
| 45 |
Because of these risks, our development efforts may not result in PRP, or any other product we attempt to develop, becoming commercially viable. If even one aspect of these development efforts is not successfully completed, required regulatory approvals will not be obtained, or if any approved products are not commercialized successfully, our business, financial condition and results of operations will be materially harmed.
A variety of factors, either alone or in concert with each other, could result in our clinical trials of PRP being delayed or unsuccessful.
While we have conducted a variety of preclinical studies, which we have concluded provide evidence to support the potential therapeutic utility of PRP, comprehensive human clinical trials in order to demonstrate the product’s safety, tolerability and efficacy will now need to be completed. Clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. A failure of one or more clinical trials can occur at any stage of testing. The outcome of preclinical testing and even early clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval of their products.
Among the numerous unforeseen events that may occur during, or as a result of, clinical trials that alone or in concert with each other could either delay or prevent our ability to receive marketing approval or commercialize PRP are the following:
| ● | regulators or institutional review boards may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site; | |
| ● | we may have delays in reaching or fail to reach an agreement on acceptable clinical trial contracts or clinical trial protocols with prospective trial sites; | |
| ● | as noted previously, clinical trials of PRP may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials or abandon product development altogether; | |
| ● | the number of patients required for clinical trials may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate, or participants may drop out of these clinical trials at a higher rate than we anticipate; | |
| ● | our third-party contractors may fail to comply with regulatory requirements or fail to meet their contractual obligations to us in a timely manner, or at all; | |
| ● | regulators or institutional review boards may require that we or our investigators suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks; | |
| ● | the cost of clinical trials may be greater than we anticipate; | |
| ● | the supply or quality of PRP or other materials necessary to conduct its clinical trials may be insufficient or inadequate; and | |
| ● | PRP may, as also noted above, have undesirable side effects or other unexpected characteristics, causing us or our investigators, regulators or institutional review boards to suspend or terminate the trials. |
| 46 |
If we are required to conduct additional clinical trials or other testing of PRP beyond those that we currently contemplate, if we are unable to successfully complete clinical trials of PRP or other testing, if the results of these trials or tests are not positive or are only modestly positive or if there are safety concerns, we may:
| ● | be delayed in obtaining marketing approval; | |
| ● | not obtain marketing approval at all; | |
| ● | obtain approval for indications or patient populations that are not as broad as intended or desired; | |
| ● | obtain approval with labeling that includes significant use or distribution restrictions or safety warnings, including boxed warnings; | |
| ● | be subject to additional post-marketing testing requirements; or | |
| ● | fail to obtain that degree of market acceptance necessary for commercial success. |
Any delay in, or termination of, our clinical trials may result in increased development costs, which would very likely cause the market price of our shares to decline and severely limit our ability to obtain additional financing and, ultimately, our ability to commercialize our products and generate product revenues. This in turn would likely materially harm our business, financial condition and operating results, and possibly lead us to cease operations.
If we fail to obtain regulatory approval in jurisdictions outside the United States, we will not be able to market PRP in those jurisdictions.
We intend to seek regulatory approval for PRP in the United Kingdom, Europe, Australia and/or other countries outside of the United States and expect that these countries will be important markets for our product, if approved. Marketing our product in these countries will require separate regulatory approvals in each market and compliance with numerous and varying regulatory requirements. The regulations that apply to the conduct of clinical trials and approval procedures vary from country to country and may require additional testing. Moreover, the time required to obtain approval may differ from that required to obtain FDA approval.
If, in the future, we are unable to establish sales and marketing capabilities or enter into agreements with third parties to sell and market PRP, we may not be successful in commercializing our product candidates if and when they are approved.
We do not have a sales or marketing infrastructure and have no experience in the sale, marketing or distribution of pharmaceutical products. To achieve commercial success for PRP or any other approved product, we must either develop a sales and marketing organization or outsource these functions to third parties. In the future, we may choose to build a focused sales and marketing infrastructure to market or co-promote some of our product candidates if and when they are approved.
There are risks involved with both establishing our own sales and marketing capabilities and entering into arrangements with third parties to perform these services. For example, recruiting and training a sales force is expensive and time consuming and could delay any product launch. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
Factors that may inhibit our efforts to commercialize our products on our own include:
| ● | our inability to recruit and retain adequate numbers of effective sales and marketing personnel; | |
| ● | the inability of sales personnel to obtain access to physicians or persuade an adequate number of physicians to prescribe any future products; | |
| ● | the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and | |
| ● | unforeseen costs and expenses associated with creating an independent sales and marketing organization. |
| 47 |
If we enter into arrangements with third parties to perform sales, marketing and distribution services, our product revenues or the profitability of these product revenues to us are likely to be lower than if we were to market and sell any products that we develop ourselves. In addition, we may not be successful in entering into arrangements with third parties to sell and market our product candidates or may be unable to do so on terms that are favorable to us. We likely will have little control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively. If we do not establish sales and marketing capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing PRP.
We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do.
The development and commercialization of new drug products is highly competitive. We face competition with respect to our current product candidate, and will face competition with respect to any product candidates that we may seek to develop or commercialize in the future from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. There are a number of large pharmaceutical and biotechnology companies that currently market and sell products or are pursuing the development of products for the treatment of the disease indications for which we are developing our product candidates. Some of these competitive products and therapies are based on scientific approaches that target and eradicate cancer stem cells to treat metastatic cancer. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
We are developing PRP for the treatment of pancreatic, ovarian and colorectal cancer. There are a variety of available therapies marketed for cancer. In many cases, these drugs are administered in combination to enhance efficacy. Some of these drugs are branded and subject to patent protection, and others are available on a generic basis. Many of these approved drugs are well-established therapies and are widely accepted by physicians, patients and third-party payors. Insurers and other third-party payors may also encourage the use of generic products. We expect that if our product candidate is approved, it will be priced at a significant premium over competitive generic products. This may make it difficult for us to achieve our business strategy of using PRP in combination with existing therapies or replacing existing therapies with PRP.
There are also a number of products in clinical development by other parties to treat and prevent metastatic cancer. Our competitors may develop products that are more effective, safer, more convenient or less costly than any that we are developing or that would render our product candidate obsolete or non-competitive. In addition, our competitors may discover biomarkers that more efficiently measure their effectiveness to treat and prevent metastatic cancer, which may give them a competitive advantage in developing potential products. Our competitors may also obtain marketing approval from the FDA or other regulatory authorities for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
Most of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller and other early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. In addition, to the extent that product or product candidates of our competitors demonstrate serious adverse side effects or are determined to be ineffective in clinical trials, the development of our product candidates could be negatively impacted.
| 48 |
Even if we are able to commercialize PRP, we will need to seek approval for reimbursement before it can be marketed, and it may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, which would harm our business.
The regulations that govern marketing approvals, pricing and reimbursement for new drug products vary widely from country to country. In the United States, recently passed legislation may significantly change the approval requirements in ways that could involve additional costs and cause delays in obtaining approvals. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for PRP in a particular country, but then be subject to price regulations that delay our commercial launch of it, possibly for lengthy time periods, and negatively impact the revenues we are able to generate from the sale of PRP in that country. Adverse pricing limitations may hinder our ability to recoup our investment in PRP, even after it has obtained marketing approval.
Our ability to commercialize PRP successfully also will depend in part on the extent to which reimbursement for it will be available from government health administration authorities, private health insurers and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels. A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. We cannot be sure that reimbursement will be available for PRP that we commercialize and, if reimbursement is available, the level of reimbursement. Reimbursement may impact the demand for, or the price of, PRP. Obtaining reimbursement for it may be particularly difficult because of the higher prices often associated with drugs administered under the supervision of a physician. If reimbursement is not available or is available only to limited levels, we may not be able to successfully commercialize PRP.
There may be significant delays in obtaining reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or similar regulatory authorities outside the United States. Moreover, eligibility for reimbursement does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies. Our inability to promptly obtain coverage and profitable payment rates from both government-funded and private payors for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
RISKS RELATED TO OUR DEPENDENCE ON THIRD PARTIES
We will depend on collaborations with third parties for the development and commercialization of PRP and other product candidates, and these collaborations may be unsuccessful.
We currently seek third-party collaborators for the development and commercialization of PRP, contract manufacturers (CMOs), contract research organizations (CROs), regulatory and development consultants, and hospitals for clinical trial sites. We intend to continue to rely on third-party collaborators for current and future product candidates for the foreseeable future. Our likely collaborators for any collaboration arrangements include large and mid-size pharmaceutical companies, regional and national pharmaceutical companies and biotechnology companies. If we do enter into any such arrangements with any third parties, we will likely have limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of our product candidates. Our ability to generate revenues from these arrangements will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements.
| 49 |
Collaborations involving our product candidates would pose the following risks to us:
| ● | collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations; | |
| ● | collaborators may not pursue development and commercialization of our product candidates or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in the collaborator’s strategic focus or available funding or external factors such as an acquisition that diverts resources or creates competing priorities; | |
| ● | collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing; | |
| ● | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products or product candidates if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours; | |
| ● | collaborators with marketing and distribution rights to one or more products may not commit sufficient resources to the marketing and distribution of such product or products; | |
| ● | collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our proprietary information or expose us to potential litigation; | |
| ● | disputes may arise between the collaborators and us that result in the delay or termination of the research, development or commercialization of our products or product candidates or that result in costly litigation or arbitration that diverts management attention and resources; and | |
| ● | collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates. |
Collaboration agreements may not lead to development or commercialization of product candidates in the most efficient manner or at all. If a present or future collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development or commercialization program could be delayed, diminished or terminated.
If we are not able to establish collaborations, we may have to alter our development and commercialization plans.
Our potential commercialization of PRP will require substantial additional cash to fund clinical trial and other expenses. As noted above, we may decide to collaborate with other pharmaceutical and biotechnology companies for the development and potential commercialization of PRP and perhaps future product candidates as well.
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA or similar regulatory authorities outside the United States, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such collaboration could be more attractive than the one with us for our product candidate. Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
| 50 |
We may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of such product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our product candidates or bring them to market and generate product revenue.
We currently contract with a third party for the manufacture of PRP and this third party may not perform satisfactorily or at all, and our reliance on any third-party for the supply of PRP carries material risks.
We do not have any manufacturing facilities or personnel. We currently obtain all of our supply of PRP for clinical development through our Manufacturing Service Agreement (the “MSA”) with Amatsigroup, and we expect to continue to rely on Amatsigroup for the manufacture of clinical and, if necessary, commercial quantities of PRP. We anticipate that our payments to Amatsigroup under the MSA will range between $2.5 million and $5.0 million over three years, when the finished drug product is manufactured and released for clinical trials. The Company has spent a total of $1,689,146 of costs to date under this contract of which $49,854 was expensed in fiscal 2019, $701,973 in fiscal 2018 and $937,319 in fiscal 2017. The MSA shall continue for a term of three years unless extended by mutual agreement in writing. Either party to the MSA has the right to terminate. The MSA expired in 2019 and may be extended by mutual agreement in writing with a possible extension currently under consideration. If we are not current with payments to Amatsigroup and Amatsigroup terminates the MSA or suspends its manufacturing services to us, this adversely affect our supply of PRP and result in harm to our business and results of operations.
This reliance on a third party increases the risk that we will not have sufficient quantities of PRP on hand at any given time, which could delay, prevent or impair our development efforts. We do not currently have alternative arrangements in place to supply us with PRP should Amatsigroup fail to perform for any reason. Amatsigroup may also fail to comply with current good manufacturing practices (“cGMP”) regulations or similar regulatory requirements outside the United States. Any such failure to comply with applicable regulations could result in sanctions being imposed on Amatsigroup, and possibly us as well. These sanctions could include fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of PRP, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect our supply of PRP and result in harm to our business and results of operations.
PRP and any other product that we may develop may compete with other product candidates and products for access to manufacturing facilities. Although we believe that there are several potential alternative manufacturers who could manufacture PRP, we may incur added costs and delays in identifying and qualifying any such replacement, as well as producing the drug product. In addition, we would then have to enter into technical transfer agreements and share our know-how with the new third-party manufacturers, which can be time-consuming and may result in delays.
Even if we were able to quickly establish agreements with other third-party manufacturers, our general reliance on third-party manufacturers entails many of the same risks as our agreement with Amatsigroup, including:
| ● | reliance on the third party for regulatory compliance and quality assurance; | |
| ● | the possible breach of the manufacturing agreement by the third party, including the misappropriation of our proprietary information, trade secrets and know-how; | |
| ● | the possible termination or nonrenewal of the agreement by the third party at a time that is costly or inconvenient for us; and | |
| ● | disruptions to the operations of our manufacturers or suppliers caused by conditions unrelated to our business or operations, including the bankruptcy of the manufacturer or supplier or a catastrophic event affecting our manufacturers or suppliers. |
| 51 |
Our current reliance on the services of Amatsigroup and current and anticipated future dependence upon others for the manufacture of PRP may adversely affect our future profit margins and our ability to commercialize any products that receive marketing approval on a timely and competitive basis.
RISKS RELATED TO OUR INTELLECTUAL PROPERTY
If we fail to comply with our obligations under any intellectual property licenses with third parties, we could lose license rights that are important to our business.
We are currently a party to a joint commercialization agreement with the University of Bath, and hope to enter into other license agreements in the future. If we fail to comply with the obligations included in any future license we may enter into in the future, such licensors may have the right to terminate these agreements, in which event we might not be able to market any product that is covered by the agreements, or to convert the exclusive licenses to non-exclusive licenses, which could materially adversely affect the value of the product candidate being developed under these license agreements. As a general matter, termination of license agreements or reduction or elimination of our licensed rights may result in our having to negotiate new or reinstated licenses with less favorable terms.
If we are unable to obtain and maintain patent protection for our technology and products, or if any licensors are unable to obtain and maintain patent protection for the technology or products that we may license from them in the future, or if the scope of the patent protection obtained is not sufficiently broad, our competitors could develop and commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and products may be adversely affected.
We have obtained patent protection for PRP in thirty-two countries and have a further thirty-three patent applications either pending or under examination in major global jurisdictions. Our future success depends in large part on our and, as applicable, our licensors’, ability to obtain and maintain patent protection in the United States and other countries with respect to our proprietary technology. We cannot be certain that patents will be issued in those countries where our applications are still under examination.
The patent process is expensive and time-consuming, and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions and has in recent years been the subject of much litigation. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are uncertain. Our pending and future patent applications may not result in patents being issued which protect our technology or products or which effectively prevent others from commercializing competitive technologies and products. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection.
The laws of foreign countries may not protect our rights to the same extent as the laws of the United States. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing. Therefore, we cannot be certain that we or our licensors were the first to make the inventions claimed in our owned or licensed patents or pending patent applications, or that we or our licensors were the first to file for patent protection of such inventions.
| 52 |
Assuming the other requirements for patentability are met, in the United States, for patents that have an effective filing date prior to March 15, 2013, the first to make the claimed invention is entitled to the patent, while outside the United States, the first to file a patent application is entitled to the patent. In March 2013, the United States transitioned to a first inventor to file system in which, assuming the other requirements for patentability are met, the first inventor to file a patent application will be entitled to the patent. We may be subject to a third party preissuance submission of prior art to the U.S. Patent and Trademark Office, or become involved in opposition, derivation, reexamination, inter parties review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights.
Even if our owned and licensed patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our owned or licensed patents by developing similar or alternative technologies or products in a non-infringing manner.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our owned and licensed patents may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
We may become involved in lawsuits to protect or enforce our patents, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours is invalid or unenforceable or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation proceeding could put one or more of our patents at risk of being invalidated or interpreted narrowly. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, our licensors may have rights to file and prosecute such claims and we are reliant on them.
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our commercial success depends upon our ability and the ability of our collaborators to develop, manufacture, market and sell PRP and any other product candidates and use our proprietary technologies without infringing the proprietary rights of third parties. We have yet to conduct comprehensive freedom-to-operate searches to determine whether our use of certain of the patent rights owned by or licensed to us would infringe patents issued to third parties. We may become party to, or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our products and technology, including interference proceedings before the U.S. Patent and Trademark Office and their European Union and global equivalents. Third parties may assert infringement claims against us based on existing patents or patents that may be granted in the future. If we are found to infringe a third party’s intellectual property rights, we could be required to obtain a license from such third party to continue developing and marketing our products and technology. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. We could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, we could be found liable for monetary damages. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could materially harm our business. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business.
| 53 |
Intellectual property litigation could cause us to spend substantial resources and distract our personnel from their normal responsibilities.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses, and could distract our personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for some of our technology and products, we also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position. We seek to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
RISKS RELATED TO REGULATORY APPROVAL OF OUR PRODUCT CANDIDATES AND OTHER LEGAL COMPLIANCE MATTERS
If we are not able to obtain, or if there are delays in obtaining, required regulatory approvals, we will not be able to commercialize PRP, and our ability to generate revenue will be materially impaired.
PRP and the activities associated with its development and commercialization, including design, testing, manufacture, safety, efficacy, recordkeeping, labeling, storage, approval, advertising, promotion, sale and distribution, are subject to comprehensive regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in other countries. Failure to obtain marketing approval for PRP will prevent us from commercializing it. We have not received approval to market PRP or any other product candidate from regulatory authorities in any jurisdiction. We have only limited experience in filing and supporting the applications necessary to gain marketing approvals and expect to rely on third-party contract research organizations to assist us in this process. Securing FDA approval requires the submission of extensive preclinical and clinical data and supporting information to the FDA for each therapeutic indication to establish PRP’s safety and efficacy. Securing FDA approval also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, the FDA. PRP may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining marketing approval or prevent or limit commercial use.
| 54 |
The process of obtaining marketing approvals, both in the United States and abroad, is expensive, may take many years if additional clinical trials are required, if approval is obtained at all, and can vary substantially based upon a variety of factors, including the type, complexity and novelty of the product candidates involved. Changes in marketing approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted product application, may cause delays in the approval or rejection of an application. The FDA has substantial discretion in the approval process and may refuse to accept any application or may decide that our data is insufficient for approval and require additional preclinical, clinical or other studies. In addition, varying interpretations of the data obtained from preclinical and clinical testing could delay, limit or prevent marketing approval of a product candidate. Any marketing approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the approved product not commercially viable.
If we experience delays in obtaining approval or if we fail to obtain approval of PRP, the commercial prospects for PRP may be harmed and our ability to generate revenues will be materially impaired.
Failure to obtain marketing approval in international jurisdictions would prevent PRP from being marketed abroad.
We intend to seek regulatory approval for PRP in a number of countries outside of the United States and expect that these countries will be important markets for it, if approved. In order to market and sell our products in the European Union, the UK, Australia and many other jurisdictions, we or our third-party collaborators must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ substantially from that required to obtain FDA approval. The regulatory approval process outside the United States generally includes all of the risks associated with obtaining FDA approval. In addition, in many countries outside the United States, it is required that the product be approved for reimbursement before the product can be approved for sale in that country. We or these third parties may not obtain approvals from regulatory authorities outside the United States on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one regulatory authority outside the United States does not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA. We may not be able to file for marketing approvals and may not receive necessary approvals to commercialize our products in any market.
PRP or any other product candidate for which we obtain marketing approval could be subject to restrictions or withdrawal from the market and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our products, when and if any of them are approved.
PRP, or any other product candidate for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data, labeling, advertising and promotional activities for such product, will be subject to continual requirements of and review by the FDA and other regulatory authorities. These requirements include submissions of safety and other post-marketing information and reports, registration and listing requirements, cGMP requirements relating to quality control, quality assurance and corresponding maintenance of records and documents, requirements regarding the distribution of samples to physicians and recordkeeping. Even if marketing approval of a product candidate is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. The FDA closely regulates the post-approval marketing and promotion of drugs to ensure drugs are marketed only for the approved indications and in accordance with the provisions of the approved labeling. The FDA imposes stringent restrictions on manufacturers’ communications regarding off-label use and if we do not market our products for their approved indications, we may be subject to enforcement action for off-label marketing.
In addition, later discovery of previously unknown problems with our products, manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may yield various results, including:
| ● | restrictions on such products, manufacturers or manufacturing processes; |
| 55 |
| ● | restrictions on the labeling or marketing of a product; | |
| ● | restrictions on product distribution or use; | |
| ● | requirements to conduct post-marketing clinical trials; | |
| ● | warning or untitled letters; | |
| ● | withdrawal of the products from the market; | |
| ● | refusal to approve pending applications or supplements to approved applications that we submit; | |
| ● | recall of products; | |
| ● | fines, restitution or disgorgement of profits or revenue; | |
| ● | suspension or withdrawal of marketing approvals; | |
| ● | refusal to permit the import or export of our products; | |
| ● | product seizure; or | |
| ● | injunctions or the imposition of civil or criminal penalties. |
Our current attempts to both expand our patent protection and seek regulatory approvals from multiple countries, as well as our future relationships with customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse and other laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings.
As we seek to obtain patent protection from multiple jurisdictions and eventually to seek marketing approval for PRP in those counties, we are and will continue to be subject to the Foreign Corrupt Practices Act, which makes it illegal for any U.S. business, even one like Propanc that is physically located in another country, to influence foreign officials with personal payments and rewards.
Moreover, healthcare providers, physicians and third-party payors will play a primary role in the recommendation and prescription of PRP and any other product candidate for which we obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our products for which we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations, include the following:
| ● | the federal healthcare anti-kickback statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under federal and state healthcare programs such as Medicare and Medicaid; | |
| ● | the federal False Claims Act imposes criminal and civil penalties, including civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; | |
| ● | the federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program and also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| 56 |
| ● | the federal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services; | |
| ● | the federal transparency requirements under the Health Care Reform Law requires manufacturers of drugs, devices, biologics and medical supplies to report to the Department of Health and Human Services information related to physician payments and other transfers of value and physician ownership and investment interests; and | |
| ● | analogous state laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers, and some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug manufacturers to report information related to payments to physicians and other health care providers or marketing expenditures. |
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines and exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any of the physicians or other providers or entities with whom we expect to do business are found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Recently enacted and future legislation, particularly in the United States, may increase the difficulty and cost for us to obtain marketing approval of and commercialize PRP and affect the prices we may obtain.
In the United States and some foreign jurisdictions there have been many legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any product candidates for which we obtain marketing approval.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (“Medicare Modernization Act”), changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for physician-administered drugs. In addition, this legislation provided authority for limiting the number of drugs that will be covered in any therapeutic class. Cost reduction initiatives and other provisions of this legislation could decrease the coverage and price that we receive for any approved products. While the Medicare Modernization Act applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. Therefore, any reduction in reimbursement that results from the Medicare Modernization Act may result in a similar reduction in payments from private payors.
In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act (“Affordable Care Act”), a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for health care and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Among other things, the Affordable Care Act revised the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states, and it imposed a significant annual fee on companies that manufacture or import branded prescription drug products.
| 57 |
At present, the future of the Affordable Care Act is the subject of significant debate in the U.S. Congress, with proposals to either partially or entirely repeal it being considered and the likelihood that there will be a new law to replace it is uncertain. It is not yet possible for us to determine the impact, if any, the enactment of any of these proposals will have on our future ability to obtain approval of or commercialize PRP.
The UK’s decision to leave the European Union could significantly increase regulatory burdens on obtaining approvals for PRP within the UK.
On March 29, 2017, the UK invoked Article 50 of Lisbon Treaty to initiate complete withdrawal from the European Union which was effected on January 31, 2020, and therefore, the regulatory drug approval process in that country may be significantly different from the current drug regulatory policies in the European Union. We currently are considering holding our clinical trials in the UK, among other countries, and therefore this event could significantly impact our efforts to successfully bring PRP to market. It is not yet possible for us to determine the impact of the UK’s withdrawal from the European Union, but any additional costs or delays in obtaining approvals may hinder our ability to conduct clinical trials or market PRP in the UK.
RISKS RELATING TO EMPLOYEE MATTERS AND MANAGING GROWTH
Our future success depends on our ability to retain our chief executive officer and our chief scientific officer and, as we continue to develop and grow as a company, to attract, retain and motivate qualified personnel.
We are highly dependent on our management team, specifically Mr. James Nathanielsz, our Chief Executive Officer, Acting Chairman, Secretary, Treasurer and a director, Carlo Campiciano, our Chief Financial Officer, and Dr. Julian Kenyon, our director who also serves as our chief scientific officer in a non-executive officer capacity. While we have a current employment agreement with Mr. Nathanielsz and a director agreement with Dr. Kenyon, both such employment agreement and director agreement permit each of the respective parties thereto to terminate such agreements upon notice. If we lose this key employee and/or the services of our other director, our business will suffer and we may have to cease operations.
Recruiting and retaining qualified scientific, clinical, manufacturing and sales and marketing personnel will also be critical to our future success, as we continue to develop PRP and attempt to grow as a company. We may not be able to attract and retain these personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors, including our scientific co-founders, may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us.
We expect to expand our development, regulatory and future sales and marketing capabilities, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
We expect to experience significant growth in the number of our employees and the scope of our operations, particularly in the areas of drug development, regulatory affairs and sales and marketing. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Due to our limited financial resources and the limited experience of our management team in managing a company with such anticipated growth, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The physical expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
| 58 |
We have identified material weaknesses in our internal control over financial reporting that, if not properly remediated, could result in material misstatements in our consolidated financial statements in future periods.
In connection with the audits of our consolidated financial statements for the fiscal years ended June 30, 2020 and 2019, and in accordance with management’s assessments of internal controls over financial reporting, we identified certain deficiencies relating to our internal control over financial reporting that constitute a material weakness under the Internal Control Integrated Framework issued by COSO in 2013. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of the company’s annual or interim financial statements will not be prevented or detected on a timely basis. A deficiency in internal control exists when the design or operation of a control does not allow management or employees, in the normal course of performing their assigned functions, to prevent or detect misstatements on a timely basis.
The following material weaknesses in our internal control over financial reporting continued to exist at June 30, 2020:
| ● | we do not have written documentation of our internal control policies and procedures. Written documentation of key internal controls over financial reporting is a requirement of Section 404 of the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”); | |
| ● | we do not have sufficient segregation of duties within accounting functions, which is a basic internal control. Due to our limited size and early stage nature of operations, segregation of all conflicting duties may not always be possible and may not be economically feasible; however, to the extent possible, the initiation of transactions, the custody of assets and the recording of transactions should be performed by separate individuals; | |
| ● | lack of independent audit committee of our board of directors; and | |
| ● | insufficient monitoring and review controls over the financial reporting closing process, including the lack of individuals with current knowledge of U.S. GAAP. |
We outsource certain functions that would normally be performed by a principal financial officer to assist us in implementing the necessary financial controls over the financial reporting and the utilization of internal management and staff to effectuate these controls.
We believe that these material weaknesses primarily relate, in part, to our lack of sufficient staff with appropriate training in U.S. GAAP and U.S. Securities and Exchange Commission (the “SEC”) rules and regulations with respect to financial reporting functions, and the lack of robust accounting systems, as well as the lack of sufficient resources to hire such staff and implement these accounting systems.
We plan to take a number of actions in the future to correct these material weaknesses including, but not limited to, establishing an audit committee of our board of directors comprised of at least two independent directors, adding additional experienced accounting and financial personnel and retaining third-party consultants to review our internal controls and recommend improvements, subject to receiving sufficient additional capital. If we receive sufficient capital, we hope to increase the chief financial officer’s role from part-time to full-time as the next step in building out our accounting department. We will need to take additional measures to fully mitigate these issues, and the measures we have taken, and expect to take, to improve our internal controls may not be sufficient to (1) address the issues identified, (2) ensure that our internal controls are effective or (3) ensure that the identified material weakness or other material weaknesses will not result in a material misstatement of our annual or interim financial statements. In addition, other material weaknesses may be identified in the future. If we are unable to correct deficiencies in internal controls in a timely manner, our ability to record, process, summarize and report financial information accurately and within the time periods specified in the rules and forms of the SEC will be adversely affected. This failure could negatively affect the market price and trading liquidity of our common stock, cause investors to lose confidence in our reported financial information, subject us to civil and criminal investigations and penalties, and generally materially and adversely impact our business and financial condition.
| 59 |
We do not have any independent directors, which represents a potential conflict of interest, and helps create a material weakness in our disclosure controls and procedures as well as our internal control over financial reporting.
We do not have any independent directors, and no audit or compensation committees that in a larger company would be expected to be comprised of independent directors. The functions of these committees, as well as other important functions that would normally be carried out by independent directors, are performed by our directors, one of whom also serves as principal executive and financial officer of the Company, resulting in an inherent and obvious conflict of interest.
Also, our lack of independent directors and an audit committee necessitates that we do not currently have a director who qualifies as an audit committee financial expert. This fact, together with our additional lack of in-house accounting personnel knowledgeable in debt and equity transactions and our extremely small administrative staff that makes it impossible to segregate critical duties, combine to create material weaknesses in both our disclosure controls and procedures and our internal control over financial reporting.
If we fail to implement and maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. In addition, any testing by us conducted in connection with Section 404 of the Sarbanes-Oxley Act, or the subsequent testing by our independent registered public accounting firm, if and when required, may reveal additional deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses or that may require prospective or retroactive changes to our consolidated financial statements or identify other areas for further attention or improvement. If in the future we identify other material weaknesses in our internal control over financial reporting, including at some of our acquired companies, if we are unable to comply with the requirements of Section 404 in a timely manner or assert that our internal control over financial reporting is effective, or if our independent registered public accounting firm is unable to express an opinion as to the effectiveness of our internal control over financial reporting, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our common stock could be negatively affected, and we could become subject to investigations by the stock exchange on which our securities are then listed, the SEC, or other regulatory authorities, which could require additional financial and management resources. Inferior internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common stock.
Additionally, we currently do not have an internal audit group nor an audit committee of our board of directors, and we will eventually need to hire additional accounting and financial staff with appropriate public company experience and technical accounting knowledge to have effective internal controls for financial reporting.
We will continue to incur significant increased costs as a result of operating as a public company.
As a public company, we will continue to incur significant legal, accounting and other expenses. For example, we are subject to mandatory reporting requirements of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), which require, among other things, that we continue to file with the SEC annual, quarterly and current reports with respect to our business and financial condition. We have incurred and will continue to incur costs associated with the preparation and filing of these SEC reports. In addition, the Sarbanes-Oxley Act, as well as rules subsequently implemented by the SEC, the Dodd-Frank Wall Street Reform and Consumer Protection Act (the “Dodd-Frank Act”) and national stock exchanges have imposed various other requirements on public companies. Stockholder activism, the current political environment and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact (in ways we cannot currently anticipate) the manner in which we operate our business. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations have and will continue to increase our legal and financial compliance costs and will make some activities more time-consuming and costly. For example, we will incur additional expense to increase our director and officer liability insurance.
| 60 |
In addition, if and when we cease to be a smaller reporting company and become subject to Section 404(b) of the Sarbanes-Oxley Act, we will be required to furnish an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with Section 404 within the prescribed time period, we will continue to be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to dedicate substantially greater internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that our independent registered public accounting firm, when required, will not be able to conclude within the prescribed timeframe that our internal control over financial reporting is effective as required by Section 404. This could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
Judgments that our stockholders obtain against us may not be enforceable.
Substantially all of our assets are located outside of the United States. In addition, our Chief Executive Officer, James Nathanielsz, and our Chief Financial Officer, Carlo Campiciano, reside in Australia and our other director, Dr. Julian Kenyon, resides in the UK. As a result, it may be difficult for you to effect service of process within the United States upon these persons. It is uncertain whether the courts of Australia or the UK would recognize or enforce judgments of the United States or state courts against us or such persons predicated upon the civil liability provisions of the laws of the United States or any state.
RISKS RELATED TO OUR COMMON STOCK
The market price of our common stock may continue to be highly volatile, you may not be able to resell your shares at or above the public offering price and you could lose all or part of your investment.
The trading price of our common stock may continue to be highly volatile. Our stock price could continue to be subject to wide fluctuations in response to a variety of factors, including the following:
| ● | actual or anticipated results of our clinical trials; | |
| ● | actions of securities analysts who initiate or maintain coverage of us, changes in financial estimates by any securities analysts who follow our company, or our failure to meet these estimates or the expectations of investors; | |
| ● | issuance of our equity and/or debt securities, or disclosure or announcements relating thereto; | |
| ● | additional shares of our common stock being sold into the market by us or our existing stockholders and/or holders of convertible debt or the anticipation of such sales; | |
| ● | stock market valuations of companies in our industry; | |
| ● | price and volume fluctuations in the overall stock market, including as a result of trends in the economy as a whole; | |
| ● | lawsuits threatened or filed against us; | |
| ● | regulatory developments in the United States and foreign countries applicable to biotech and biopharma companies; and | |
| ● | other events or factors, including those resulting from war or incidents of terrorism, or responses to these events. |
| 61 |
The stock markets in general, and the small-cap biotech market, in particular, have experienced extreme price and volume fluctuations in recent years that have significantly affected the quoted prices of the securities of many companies, including companies in our industry. The changes often appear to occur without regard to specific operating performance. The price of our shares of common stock could fluctuate based upon factors that have little or nothing to do with our company and these fluctuations could materially reduce our share price. Broad market, clinical trial results and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance.
Currently there is a limited public market for our common stock, and we cannot predict the future prices or the amount of liquidity of our common stock.
Currently, there is a limited public market for our common stock. Our common stock is quoted on the OTCQB under the symbol “PPCB.” However, the OTCQB is not a liquid market in contrast to the major stock exchanges. We cannot assure you as to the liquidity or the future market prices of our common stock if a market does develop. If an active market for our common stock does not develop, the fair market value of our common stock could be materially adversely affected. We cannot predict the future prices of our common stock.
The designation of our common stock as a “penny stock” would limit the liquidity of our common stock.
Our common stock may be deemed a “penny stock” (as that term is defined under Rule 3a51-1 of the Exchange Act) in any market that may develop in the future. Generally, a “penny stock” is a common stock that is not listed on a securities exchange and trades for less than $5.00 a share. Prices often are not available to buyers and sellers and the market may be very limited. Broker-dealers who sell penny stocks must provide purchasers of these stocks with a standardized risk-disclosure document prepared by the SEC. The document provides information about penny stocks and the nature and level of risks involved in investing in the penny stock market. A broker must also provide purchasers with bid and offer quotations and information regarding broker and salesperson compensation and make a written determination that the penny stock is a suitable investment for the purchaser and obtain the purchaser’s written agreement to the purchase. Many brokers choose not to participate in penny stock transactions. Because of the penny stock rules, there may be less trading activity in any market that develops for our common stock in the future and stockholders are likely to have difficulty selling their shares.
Although our common stock is currently quoted on the OTCQB, if we do not meet or comply with the OTCQB’s quotation requirements, our shares would be downgraded from the OTCQB and would be traded on the OTC Pink (aka the Pink Sheets).
Although our common stock is currently quoted on the OTCQB, to be eligible to continue to be quoted on the OTCQB, among other things, our common stock is required to meet a minimum closing bid test of $0.01 per share on at least one of the prior thirty consecutive calendar days. On July 2, 2020, the OTCQB informed us that the bid price for our common stock has closed below $0.01 for more than 30 consecutive calendar days, and therefore, our common stock no longer meets the Standards for Continued Eligibility for OTCQB. The OTCQB has granted us a cure period of 90 calendar days from such date (being September 29, 2020) during which the minimum closing bid price of our common stock must be $0.01 or greater for ten consecutive trading days in order for our common stock to continue trading on the OTCQB.
Our management determined that a Reverse Stock Split would allow us to satisfy such minimum closing price requirement for the duration of the required period. On August 22, 2020 we submitted a Form 14C to Finra to affect a 1:1000 reverse split of the Company’s stock. The application has progressed and there are no matters outstanding that have not been addressed by the Company and as such we believe that the application is in the final stages of approval by Finra. At the date of this report the reverse split has not been affected and the Company’s stock has been removed from the OTCQB marketplace and moved to the Pink market. The Company will reapply for quotation on the OTCQB market as soon as the reverse split is affected.
| 62 |
Because our directors and officers currently and for the foreseeable future will continue to control our Company, it is not likely that you will be able to elect directors or have any say in the policies of our Company.
Our stockholders are not entitled to cumulative voting rights. Consequently, the election of directors and all other matters requiring stockholder approval will be decided by majority vote. Our directors and officers beneficially own less than 1.0% of our outstanding common stock. In addition, our chief executive officer and chief financial officer beneficially owns all of our preferred stock, which entitles him, as a holder of Series A preferred stock, to vote on all matters submitted or required to be submitted to a vote of the stockholders, except election and removal of directors, and each share entitles him to five hundred votes per share of Series A preferred stock, and as a holder of Series B preferred stock, to voting power equivalent of the number of votes equal to the total number of shares of common stock outstanding as of the record date for the determination of stockholders entitled to vote at each meeting of our stockholders and entitled to vote on all matters submitted or required to be submitted to a vote of our stockholders. Due to such a disproportionate voting power, new investors will not be able to affect a change in our business or management, and therefore, stockholders would have limited recourse as a result of decisions made by management.
Moreover, this preferred stock ownership may discourage a potential acquirer from making a tender offer or otherwise attempting to obtain control of us, which in turn could reduce our stock price or prevent our stockholders from realizing a premium over our stock price.
Future sales and issuances of our common stock or rights to purchase common stock could result in additional dilution of the percentage ownership of our stockholders and could cause our stock price to decline.
We are authorized to issue up to 1,000,000,000 shares of our common stock, $0.001 par value per share. We have the right to raise additional capital or incur borrowings from third parties to finance our business. The board of directors has the authority, without the consent of any of the shareholders, to cause us to issue more shares of our common stock and/or securities convertible into our common stock. We will likely issue additional shares of our common stock and/or such securities in the future and such future sales and issuances of our common stock or rights to purchase our common stock could result in substantial dilution to our existing stockholders. We may sell common stock, convertible securities and other equity securities in one or more transactions at prices and in a manner as we may determine from time to time. If we sell any such securities in subsequent transactions, our stockholders may be materially diluted. New investors in such subsequent transactions could gain rights, preferences and privileges senior to those of holders of our common stock.
In the future, we may issue additional preferred stock without the approval of our stockholders, which could make it more difficult for a third party to acquire us and could depress our stock price.
We are authorized to issue up to 1,500,005 shares of our preferred stock, par value $0.01 per share, having such rights, preferences and privileges as are determined by our board of directors in their discretion. We have the right to raise additional capital or incur borrowings from third parties to finance our business. The board of directors has the authority, without the consent of any of the stockholders, to cause us to issue more shares of our preferred stock. Our board of directors may issue, and has in the past issued, without a vote of our stockholders, one or more series of our preferred stock with such rights and preferences as it determines. This could permit our board of directors to issue preferred stock to investors who support us and our management and permit our management to retain control of our business. Additionally, issuance of preferred stock could block an acquisition which could result in both a drop in our stock price and a decline in interest of our common stock.
Since we intend to retain any earnings for development of our business for the foreseeable future, you will likely not receive any dividends for the foreseeable future, and capital appreciation, if any, will be the source of gain for our stockholders.
We have never declared or paid any cash dividends or distributions on our capital stock. We currently intend to retain our future earnings to support operations and to finance expansion and therefore we do not anticipate paying any cash dividends on our common stock in the foreseeable future. As a result, capital appreciation, if any, of our common stock will be the sole source of gain for our stockholders for the foreseeable future.
| 63 |
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
Section 382 (“Section 382”) of the Internal Revenue Code of 1986, as amended (the “Code”), contains rules that limit the ability of a company that undergoes an ownership change to utilize its net operating losses (“NOLs”) and tax credits existing as of the date of such ownership change. Under the rules, such an ownership change is generally any change in ownership of more than 50% of a company’s stock within a rolling three-year period. The rules generally operate by focusing on changes in ownership among stockholders considered by the rules as owning, directly or indirectly, 5% or more of the stock of a company and any change in ownership arising from new issuances of stock by the company. As a result of this Section 382 limitation, any ownership changes as defined by Section 382 may limit the amount of NOL carryforwards that could be utilized annually to offset future taxable income.
As a smaller reporting company, we are subject to scaled disclosure requirements that may make it more challenging for investors to analyze our results of operations and financial prospects.
As a “smaller reporting company,” we (i) are able to provide simplified executive compensation disclosures in our filings, (ii) are exempt from the provisions of Section 404(b) of the Sarbanes-Oxley Act requiring that independent registered public accounting firms provide an attestation report on the effectiveness of internal control over financial reporting and (iii) have certain other decreased disclosure obligations in our filings with the SEC, including being required to provide only two years of audited financial statements in annual reports. Consequently, it may be more challenging for investors to analyze our results of operations and financial prospects.
We will remain a smaller reporting company until the beginning of a fiscal year in which we had a public float of $250 million held by non-affiliates as of the last business day of the second quarter of the prior fiscal year, assuming our common stock is registered under Section 12 of the Exchange Act on the applicable evaluation date. Even if we remain a smaller reporting company, if our public float exceeds $250 million and our annual revenues are greater than $100 million, we will become subject to the provisions of Section 404(b) of the Sarbanes-Oxley Act.
***
The risks above do not necessarily comprise of all those associated with an investment in our Company. This Registration Statement contains forward looking statements that involve unknown risks, uncertainties and other factors that may cause our actual results, financial condition, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by such forward looking statements. Factors that might cause such a difference include, but are not limited to, those set out above.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus may contain certain “forward-looking” statements as such term is defined by the SEC in its rules, regulations and releases, which represent the registrant’s expectations or beliefs, including but not limited to, statements concerning our operations, economic performance, financial condition, growth and acquisition strategies, investments, and future operational plans. For this purpose, any statements contained herein that are not statements of historical fact may be deemed to be forward-looking statements. Without limiting the generality of the foregoing, words such as “may,” “will,” “expect,” “believe,” “anticipate,” “intent,” “could,” “would,” “should,” “estimate,” “might,” “plan,” “predict” or “continue” or the negative or other variations thereof or comparable terminology are intended to identify forward-looking statements. These statements by their nature involve substantial risks and uncertainties, certain of which are beyond our control, and actual results may differ materially depending on a variety of important factors, including uncertainty related to the discovery, development and commercialization of our product candidate, protection of our intellectual property, governmental regulation, the operations of our Company and our subsidiaries, managing and maintaining growth, volatility of our stock price, and any other factors discussed in this and our other filings with the SEC.
These risks and uncertainties and other factors include, but are not limited to those set forth under the section captioned “Risk Factors” of this prospectus. Given these risks and uncertainties, readers are cautioned not to place undue reliance on our forward-looking statements. All subsequent written and oral forward-looking statements attributable to us or to persons acting on our behalf are expressly qualified in their entirety by these cautionary statements. Except as otherwise required by applicable law, we undertake no obligation to publicly update or revise any forward-looking statements or the risk factors described in this prospectus or in the documents we incorporate by reference, whether as a result of new information, future events, changed circumstances or any other reason after the date of this prospectus.
| 64 |
This prospectus contains forward-looking statements, including statements regarding, among other things:
| ● | our ability to continue as a going concern; | |
| ● | our anticipated needs for working capital; | |
| ● | our ability to successfully develop PRP, our lead product candidate; | |
| ● | our ability to reach research and development milestones as planned and within proposed budgets; | |
| ● | our current reliance on substantial debt financing; | |
| ● | our ability to repay current debt in cash and obtain adequate new financing; | |
| ● | our dependence on third parties for services; | |
| ● | our dependence on key executives; | |
| ● | our ability to control costs; | |
| ● | our ability to successfully implement our expansion strategies; | |
| ● | our ability to successfully develop and market our technologies; | |
| ● | our ability to obtain and maintain patent protection; | |
| ● | our ability to recruit employees with regulatory, accounting and finance expertise; | |
| ● | the impact of government regulations, including United States Food and Drug Administration (the “FDA”) regulations; | |
| ● | the impact of any future litigation; | |
| ● | the availability of capital; and | |
| ● | changes in economic, business and competitive conditions; |
Actual events or results may differ materially from those discussed in forward-looking statements as a result of various factors, including, without limitation, the risks, uncertainties and other factors outlined in the section captioned “Risk Factors” of this prospectus and matters described in this prospectus generally. In light of these risks and uncertainties, there can be no assurance that the forward-looking statements contained in this prospectus will in fact occur. We caution you not to place undue reliance on these forward-looking statements. In addition to the information expressly required to be included in this prospectus, we will provide such further material information, if any, as may be necessary to make the required statements, in light of the circumstances under which they are made, not misleading. All subsequent written and oral forward-looking statements attributable to our Company or to persons acting on our behalf are expressly qualified in their entirety by these cautionary statements. Except as required by law, we do not intend to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise.
| 65 |
This Prospectus relates to shares of our common stock that may be offered and sold from time to time by the Selling Stockholder. We will receive no proceeds from the sale of shares of common stock by the Selling Stockholder in this registration statement. The proceeds from the sales will belong to the Selling Stockholder. However, we will receive proceeds from the sale of the Purchase Shares to the Selling Stockholder pursuant to the Common Stock Purchase Agreement.
DETERMINATION OF OFFERING PRICE
The prices at which the shares of common stock are covered by this prospectus may actually be sold will be determined by the prevailing public market price for shares of our common stock, by negotiations between the Selling Shareholder and buyers of our common stock in private transactions or as otherwise described in “Plan of Distribution.”
The common stock being offered by the Selling Stockholder are those previously issued to the Selling Stockholder, and those issuable to the selling shareholders, upon exercise of the warrants. We are registering the shares of common stock in order to permit the Selling Stockholder to offer the shares for resale from time to time. Except for the ownership of the shares of common stock and the warrants, the Selling Stockholder has not had any material relationship with us within the past three years.
The table below lists the Selling Stockholder and other information regarding the beneficial ownership of the shares of common stock by the Selling Stockholder. The second column lists the number of shares of common stock beneficially owned by the Selling Stockholder, based on its ownership of the shares of common stock and warrants, as of October 13, 2020, assuming exercise of the warrants held by the Selling Stockholder on that date, without regard to any limitations on exercises.
The third column lists the shares of common stock being offered by this prospectus by the Selling Stockholder.
| 66 |
In accordance with the terms of a registration rights agreement with the Selling Stockholder, this prospectus generally covers the resale of the sum of (i) the number of shares of common stock issued to the selling shareholders and (ii) the maximum number of shares of common stock issuable upon exercise of the related warrants, determined as if the outstanding warrants were exercised in full as of the trading day immediately preceding the date this registration statement was initially filed with the SEC, each as of the trading day immediately preceding the applicable date of determination and all subject to adjustment as provided in the registration right agreement, without regard to any limitations on the exercise of the warrants. The fourth column assumes the sale of all of the shares offered by the Selling Stockholder pursuant to this prospectus.
Under the terms of the warrants, the Selling Stockholder may not exercise the warrants to the extent such exercise would cause the Selling Stockholder, together with its affiliates and attribution parties, to beneficially own a number of shares of common stock which would exceed 4.99% of our then outstanding common stock following such exercise, excluding for purposes of such determination shares of common stock issuable upon exercise of the warrants which have not been exercised. The number of shares in the second column does not reflect this limitation. The Selling Stockholder may sell all, some or none of their shares in this offering. See “Plan of Distribution.”
| Name of Selling Stockholder | Number of shares of Common Stock Owned Prior to Offering | Maximum Number of shares of Common Stock to be Sold Pursuant to this Prospectus | Number of shares of Common Stock Owned After Offering (1)(2) | |||||||||
| Ionic Ventures LLC (3) | 0 | 150,000,000 | 0 | |||||||||
(1) Includes shares of Common Stock underlying the Warrants that may held by the Selling Stockholder that are covered by this prospectus, including any such securities that, due to contractual restrictions, may not be exercisable if such conversion would result in beneficial ownership greater than 4.99%.
(2) Assumes that the Selling Stockholder sells all of the common stock underlying the Pre-funded Warrants and Warrants offered pursuant to this prospectus.
(3) Ionic Ventures LLC is controlled by Brendan O’Neill, whose address is 5328 Yacht Haven Grande Box # 15 Suite C201, St. Thomas, VI 00802.
The Selling Stockholder of the securities and any of its pledgees, assignees and successors-in-interest may, from time to time, sell any or all of their securities covered hereby on the principal trading market or any other stock exchange, market or trading facility on which the securities are traded or in private transactions. These sales may be at fixed or negotiated prices. The Selling Stockholder may use any one or more of the following methods when selling securities:
| ● | ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers; | |
| ● | block trades in which the broker-dealer will attempt to sell the securities as agent but may position and resell a portion of the block as principal to facilitate the transaction; | |
| ● | purchases by a broker-dealer as principal and resale by the broker-dealer for its account; | |
| ● | an exchange distribution in accordance with the rules of the applicable exchange; | |
| ● | privately negotiated transactions; | |
| ● | settlement of short sales; | |
| ● | in transactions through broker-dealers that agree with the Selling Stockholder to sell a specified number of such securities at a stipulated price per security; | |
| ● | through the writing or settlement of options or other hedging transactions, whether through an options exchange or otherwise; |
| 67 |
| ● | a combination of any such methods of sale; or | |
| ● | any other method permitted pursuant to applicable law. |
The Selling Stockholder may also sell securities under Rule 144 or any other exemption from registration under the Securities Act of 1933, as amended (the “Securities Act”), if available, rather than under this prospectus.
Broker-dealers engaged by the Selling Stockholders may arrange for other brokers-dealers to participate in sales. Broker-dealers may receive commissions or discounts from the Selling Stockholders (or, if any broker-dealer acts as agent for the purchaser of securities, from the purchaser) in amounts to be negotiated, but, except as set forth in a supplement to this Prospectus, in the case of an agency transaction not in excess of a customary brokerage commission in compliance with FINRA Rule 2440; and in the case of a principal transaction a markup or markdown in compliance with FINRA IM-2440.
In connection with the sale of the securities or interests therein, the Selling Stockholders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the securities in the course of hedging the positions they assume. The Selling Stockholders may also sell securities short and deliver these securities to close out their short positions, or loan or pledge the securities to broker-dealers that in turn may sell these securities. The Selling Stockholders may also enter into option or other transactions with broker-dealers or other financial institutions or create one or more derivative securities which require the delivery to such broker-dealer or other financial institution of securities offered by this prospectus, which securities such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).
The Selling Stockholders and any broker-dealers or agents that are involved in selling the securities may be deemed to be “underwriters” within the meaning of the Securities Act in connection with such sales. In such event, any commissions received by such broker-dealers or agents and any profit on the resale of the securities purchased by them may be deemed to be underwriting commissions or discounts under the Securities Act. Each Selling Stockholder has informed the Company that it does not have any written or oral agreement or understanding, directly or indirectly, with any person to distribute the securities.
The Company is required to pay certain fees and expenses incurred by the Company incident to the registration of the securities. The Company has agreed to indemnify the Selling Stockholders against certain losses, claims, damages and liabilities, including liabilities under the Securities Act.
We agreed to keep this prospectus effective until the earlier of (i) the date on which the securities may be resold by the Selling Stockholders without registration and without regard to any volume or manner-of-sale limitations by reason of Rule 144, without the requirement for the Company to be in compliance with the current public information under Rule 144 under the Securities Act or any other rule of similar effect or (ii) all of the securities have been sold pursuant to this prospectus or Rule 144 under the Securities Act or any other rule of similar effect. The resale securities will be sold only through registered or licensed brokers or dealers if required under applicable state securities laws. In addition, in certain states, the resale securities covered hereby may not be sold unless they have been registered or qualified for sale in the applicable state or an exemption from the registration or qualification requirement is available and is complied with.
Under applicable rules and regulations under the Exchange Act, any person engaged in the distribution of the resale securities may not simultaneously engage in market making activities with respect to the common stock for the applicable restricted period, as defined in Regulation M, prior to the commencement of the distribution. In addition, the Selling Stockholders will be subject to applicable provisions of the Exchange Act and the rules and regulations thereunder, including Regulation M, which may limit the timing of purchases and sales of the common stock by the Selling Stockholders or any other person. We will make copies of this prospectus available to the Selling Stockholders and have informed them of the need to deliver a copy of this prospectus to each purchaser at or prior to the time of the sale (including by compliance with Rule 172 under the Securities Act).
| 68 |
MARKET PRICE OF AND DIVIDENDS ON OUR COMMON EQUITY AND RELATED STOCKHOLDER MATTERS
Market Information
Our common stock is quoted under the trading symbol “PPCB” on the OTC Markets - Pink. Only a limited market exists for our common stock. There is no assurance that a regular trading market will develop, or if developed, that it will be sustained. Therefore, a stockholder may be unable to resell his securities in our Company.
The following table sets forth the range of high and low closing prices for our common stock for each of the periods indicated as reported by the OTCQB. These quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission and may not necessarily represent actual transactions.
High ($) | Low ($) | |||||||
| Fourth quarter ended June 30, 2020 | $ |
.029 | † | $ | 0.0037 | |||
| Third quarter ended March 31, 2020 | $ | 0.75 | † | $ | 0.03 | † | ||
| Second quarter ended December 31, 2019 | $ | 0.98 | † | $ | 0.11 | † | ||
| First quarter ended September 30, 2019 | $ | 6.49 | † | $ | 0.75 | † | ||
| Fiscal Year Ended June 30, 2019 | ||||||||
| Fourth quarter ended June 30, 2019 | $ | 6.3 | † | $ | 0.06 | † | ||
| Third quarter ended March 31, 2019 | $ | 15 | * | $ | 5 | * | ||
| Second quarter ended December 31, 2018 | $ | 50 | * | $ | 6 | * | ||
| First quarter ended September 30, 2018 | $ | 144.4 | * | $ | 1.95 | * | ||
| Fiscal Year Ended June 30, 2018 | ||||||||
| Fourth quarter ended June 30, 2018 | $ | 50 | * | $ | 21 | * | ||
| Third quarter ended March 31, 2018 | $ | 125 | * | $ | 44.55 | * | ||
| Second quarter ended December 31, 2017 | $ | 385 | * | $ | 45 | * | ||
| First quarter ended September 30, 2017 | $ | 550 | * | $ | 115 | * | ||
† Reflects 1-for-500 reverse stock split
* Does not reflect 1-for-500 reverse stock split
On October 7, 2020 the last reported sales price per share of our common stock on the OTC Markets - Pink was $0.0023.
Number of Holders
As of September 28, 2020, we had 74 record holders of our common stock holding 688,670,618 shares, one holder of our Series A Preferred Stock holding 500,000 shares and one holder of our Series B Preferred Stock holding one share.
Penny Stock
The SEC has adopted rules that regulate broker-dealer practices in connection with transactions in penny stocks. Penny stocks are generally equity securities with a market price of less than $5.00, other than securities registered on certain national securities exchanges or quoted on the NASDAQ system, provided that current price and volume information with respect to transactions in such securities is provided by the exchange or system. The penny stock rules require a broker-dealer, prior to a transaction in a penny stock, to deliver a standardized risk disclosure document prepared by the SEC, that: (a) contains a description of the nature and level of risk in the market for penny stocks in both public offerings and secondary trading; (b) contains a description of the broker’s or dealer’s duties to the customer and of the rights and remedies available to the customer with respect to a violation of such duties or other requirements of the securities laws; (c) contains a brief, clear, narrative description of a dealer market, including bid and ask prices for penny stocks and the significance of the spread between the bid and ask price; (d) contains a toll-free telephone number for inquiries on disciplinary actions; (e) defines significant terms in the disclosure document or in the conduct of trading in penny stocks; and (f) contains such other information and is in such form, including language, type size and format, as the SEC shall require by rule or regulation.
| 69 |
The broker-dealer also must provide, prior to effecting any transaction in a penny stock, the customer with (a) bid and offer quotations for the penny stock; (b) the compensation of the broker-dealer and its salesperson in the transaction; (c) the number of shares to which such bid and ask prices apply, or other comparable information relating to the depth and liquidity of the market for such stock; and (d) a monthly account statement showing the market value of each penny stock held in the customer’s account.
In addition, the penny stock rules require that prior to a transaction in a penny stock not otherwise exempt from those rules, the broker-dealer must make a special written determination that the penny stock is a suitable investment for the purchaser and receive the purchaser’s written acknowledgment of the receipt of a risk disclosure statement, a written agreement as to transactions involving penny stocks, and a signed and dated copy of a written suitability statement.
These disclosure requirements may have the effect of reducing the trading activity for our common stock. Therefore, stockholders may have difficulty selling our securities.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table shows the number of shares of our common stock that we have issued under our equity compensation plans as of October 13, 2020.
| Plan Category | Number
of Securities to be Issued Upon Exercise of Outstanding Options | Weighted Average Exercise Price of Outstanding Options | Number
of Securities Remaining Available for Future Issuance Under Equity Compensation Plans | |||||||||
| Equity Compensation Plans Not Approved by Security Holders | 59,644 | (1)(2)(3) | $ | 76.37 | 174,356 | (2) | ||||||
| Total | 59,644 | (1)(2)(3) | $ | 76.37 | 174,356 | (2) | ||||||
| (1) | On April 14, 2016, our board of directors granted options to purchase shares of our common stock to each of James Nathanielsz, our Chief Executive Officer, acting Chief Financial Officer (through June 30, 2019) and a director, and Dr. Julian Kenyon, our director. We granted 572 stock options at an exercise price of $3,750 per share (market value of our shares on the grant date), to each of Mr. Nathanielsz and Mr. Kenyon. 191 of such options vested on April 14, 2016 and expire on April 14, 2021, 191 of such options vested on April 14, 2017 (first anniversary of the grant date) and expire on April 14, 2021, and 191 of such options vested on April 14, 2018 (second anniversary of the grant date) and expire on April 14, 2021. The fair value of each of the 572 options at the grant date was $1,962,440 (aggregate total of $3,924,880). | |
| (2) | On May 14, 2019, our board of directors granted options to purchase shares of our common stock to each of James Nathanielsz, our Chief Executive Officer, acting Chief Financial Officer (through June 30, 2019) and a director, and Dr. Julian Kenyon, our director. We granted stock options to purchase 39,000 and 19,500 shares of the Company’s common stock to the Company’s Chief Executive Officer and Chief Scientific Officer, respectively. The total 58,500 options have a term of 10 years from the date of grant and exercise price ranging from $4.25 to $4.675 per share. 1/3rd of these options shall vest every successive one-year anniversary, provided, that on each such vesting date, the Chief Executive Officer and Chief Scientific Officer are employed by the Company and subject to the other provisions of the employment agreement. The 58,500 stock options were valued using a Black-Scholes model with the following assumptions: stock price at valuation date of $4.25 based on quoted trading price on date of grant, exercise price of $4.65, dividend yield of zero, years to maturity of 10.00, a risk free rate of 2.42%, and expected volatility 268% for a total value of $248,620. | |
| (3) | Mr. Nathanielsz and Dr. Kenyon have the option under their individual employment and director agreements, respectively, to convert any accrued but unpaid salary or fees, as the case may be, into shares of our common stock at a conversion rate between par value and the closing bid price on the date of conversion to be determined by the parties. |
2019 Equity Incentive Plan
On May 14, 2019, our board of directors adopted our 2019 Equity Incentive Plan (the “2019 Plan”), which reserves a total of 234,000 shares of our common stock for issuance under the 2019 Plan. As described below, incentive awards authorized under the 2019 Plan include, but are not limited to, incentive stock options within the meaning of Section 422 of the Internal Revenue Code of 1986, as amended (the “Code”). If an incentive award granted under the 2019 Plan expires, terminates, is unexercised or is forfeited, or if any shares are surrendered to us in connection with the exercise of an incentive award, the shares subject to such award and the surrendered shares will become available for further awards under the 2019 Plan.
| 70 |
Administration ― Our board of directors will administer the 2019 Plan. Subject to the terms of the 2019 Plan, our board of directors has complete authority and discretion to determine the terms upon which awards may be granted under the 2019 Plan.
Grants ― The 2019 Plan authorizes the grant to participants of nonqualified stock options, incentive stock options, restricted stock awards, restricted stock units, performance grants intended to comply with Section 162(m) of the Code and stock appreciation rights, as described below:
| ● | Options granted under the 2019 Plan entitle the grantee, upon exercise, to purchase up to a specified number of shares from us at a specified exercise price per share. The exercise price for shares of Common Stock covered by an option generally cannot be less than the fair market value of Common Stock on the date of grant unless agreed to otherwise at the time of the grant. In addition, in the case of an incentive stock option granted to an employee who, at the time the incentive stock option is granted, owns stock representing more than 10% of the voting power of all classes of stock of the Company or any parent or subsidiary, the per share exercise price will be no less than 110% of the fair market value of Common Stock on the date of grant. | |
| ● | Restricted stock awards and restricted stock units may be awarded on terms and conditions established by the compensation committee, which may include performance conditions for restricted stock awards and the lapse of restrictions on the achievement of one or more performance goals for restricted stock units. | |
| ● | The board of directors may make performance grants, each of which will contain performance goals for the award, including the performance criteria, the target and maximum amounts payable, and other terms and conditions. | |
| ● | The 2019 Plan authorizes the granting of stock awards. The board of directors will establish the number of shares of our common stock to be awarded (subject to the aggregate limit established under the 2019 Plan upon the number of shares of our common stock that may be awarded or sold under the 2019 Plan) and the terms applicable to each award, including performance restrictions. | |
| ● | Stock appreciation rights (“SARs”) entitle the participant to receive a distribution in an amount not to exceed the number of shares of Common Stock subject to the portion of the SAR exercised multiplied by the difference between the market price of a share of Common Stock on the date of exercise of the SAR and the market price of a share of our common Stock on the date of grant of the SAR. |
Duration, Amendment, and Termination ― Our board of directors has the power to amend, suspend or terminate the 2019 Plan without stockholder approval or ratification at any time or from time to time. No change may be made that increases the total number of shares of Common Stock reserved for issuance pursuant to incentive awards or reduces the minimum exercise price for options or exchange of options for other incentive awards, unless such change is authorized by our stockholders within one year of such change. Unless sooner terminated, the 2019 Plan would terminate ten years after it is adopted.
No awards or any shares of our common stock were issued during the fiscal year 2020 under the 2019 Plan.
DIVIDEND POLICY
We have not paid any cash dividends to our stockholders. The declaration of any future cash dividends is at the discretion of our Board and depends upon our earnings, if any, our capital requirements and financial position, and general economic conditions. It is our present intention not to pay any cash dividends in the foreseeable future, but rather to reinvest earnings, if any, in our business operations.
SELECTED FINANCIAL DATA
Not required for smaller reporting companies.
| 71 |
DILUTION
Not applicable. The shares registered under this registration statement are not being offered for purchase by the Company. The shares are being registered on behalf of the Selling Stockholder (the Selling Stockholder identified in this prospectus).
| 72 |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and operating results together with our consolidated financial statements and related notes included elsewhere in this prospectus. This discussion and analysis and other parts of this prospectus contain forward-looking statements based upon current beliefs, plans and expectations that involve risks, uncertainties and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of various factors, including those set forth under “Risk Factors” or in other parts of this prospectus. The last day of our fiscal year is June 30. Our fiscal quarters end on September 30, December 31, March 31 and June 30 and our current fiscal year ends on June 30, 2021. U.S. Dollars are denoted herein by “USD,” “$” and “dollars”.
Overview
We were incorporated in the state of Delaware as Propanc Health Group Corporation on November 23, 2010. In January 2011, to reorganize our Company, we acquired all of the outstanding shares of Propanc PTY LTD, an Australian corporation, on a one-for-one basis and Propanc PTY LTD became our wholly-owned subsidiary. Effective April 20, 2017, we changed our name to “Propanc Biopharma, Inc.” to better reflect our current stage of operations and development.
We are a development-stage healthcare company that is currently focused on developing new cancer treatments for patients suffering from pancreatic, ovarian and colorectal cancer. Utilizing our scientific and oncology consultants, we have developed a rational, composite formulation of anti-cancer compounds, which together exert a number of effects designed to control or prevent tumors from recurring and spreading through the body. Our lead product candidate, PRP, is a variation upon our novel formulation and involves pro-enzymes, the inactive precursors of enzymes.
As a result of positive early indications of the anti-cancer effects of our technology, over the last 12-18 months we have conducted successful pre-clinical studies on PRP. Subject to us receiving sufficient financing, we plan to begin our Investigational Medicinal Product Dossier, study proposal and Investigator’s Brochure in the second half of 2020 calendar year. Our plan is to then commence our study preparation process with the contract research organization, analytical lab and trial site(s) selection and to begin our clinical trial application for PRP (“CTA”) compilation in the third calendar quarter of 2020, and complete the CTA compilation and submit the CTA in the fourth calendar quarter of 2020. In the second quarter of 2021, we plan to begin the preparation of logistics and trial site initiation visits. Subject to raising additional sufficient capital, we subsequently plan to commence a First-In-Human (FIH), Phase Ib study in patients with advanced solid tumors, evaluating the safety, pharmacokinetics and anti-tumor efficacy of PRP in the fourth calendar quarter of 2020, which study we hope to complete within twelve months thereafter. We intend to develop our PRP to treat early-stage cancer and pre-cancerous diseases and as a preventative measure for patients at risk of developing cancer based on genetic screening.
To date, we have generated no revenue, have no cancer treatment products available to market and have no products which have reached the clinical trial stage. We require substantial additional financing to continue to test and commercialize PRP.
| 73 |
Recent Developments
Business Developments
In July 2019, we appointed Mr. Carlo Campiciano as Chief Financial Officer. Mr. Campiciano brings significant experience to the Company across a broad range of financial disciplines in the healthcare sector, including taxation, finance, operations, planning and financial strategy. Mr. Campiciano will assist with the transformational stage into a clinical development company, along with the goal of up-listing to a U.S. national stock exchange.
In January 2020, we announced that a Certificate for Advance Overseas Finding was received from the Board of Innovation and Science Australia to receive up to a 43.5% “cash back” benefit from overseas R&D expenses. The finding relates to the planned Phase 1 clinical trial – Multiple Ascending Dose Studies of proteolytic proenzymes for the treatment of pancreatic cancer. Overseas activities to be undertaken include the development of an analytical assay for the quantification of active pharmaceutical ingredients in the Company’s lead product candidate, PRP, and its manufacture of the finished product for the Phase 1 clinical trial.
In November 2019, we announced today that our POP1 research and drug discovery program has made significant advancements towards producing synthetic versions of the two proenzymes, trypsinogen and chymotrypsinogen. With the aim of producing large quantities of trypsinogen and chymotrypsinogen for commercial use, exhibiting minimal variation between lots and without sourcing the proenzymes from animals, the Company is undertaking a challenging research project in collaboration with the universities of Jaén and Granada. The two active ingredients are currently naturally derived from animal sources, which combine to form our lead product candidate, PRP. Our vision is to produce a backup product candidate to PRP which can further stabilize and enhance the effects of the proenzymes when administered to patients. At the research laboratories at the universities of Jaén and Granada, scientific researchers are in the process of optimizing conditions to achieve high titers of recombinant trypsinogen and chymotrypsinogen with this expression system.
In August 2019, we announced that we have developed a method to quantify the active ingredients of our lead product candidate, PRP, in preparation for the company’s First-In-Human (“FIH”) study, planned for 2020. The work was conducted by Propanc’s research partner based in Berlin, Germany, who has extensive experience in the development of functional assays for unique bio-therapeutics. This bioanalytical method development and validation plays a significant role in evaluation and interpretation of the systemic absorption of PRP in clinical studies including its distribution, and clinical effects throughout the body. The development of the bioanalytical assay is also an important step for the clinical development of PRP, as Propanc evaluates sites to conduct the FIH study in advanced cancer patients, such as the Peter Mac Center, Australia’s largest cancer hospital, which has significant experience in early stage clinical development. Validation of the bioanalytical method will be undertaken in 2020.
In March 2019, we announced that we have initiated development of a bio-analytical assay intended to quantify the active ingredients of our lead product candidate, PRP, in preparation for human trials, planned for the beginning of the 2020 calendar year. The work will be conducted by a specialist Contract Research Organization with extensive knowledge in the development of functional assays for different bio-therapeutics. PRP is a solution of two proenzymes, trypsinogen and chymotrypsinogen, administered by I.V. injection. Development of the bio-analytical assay will be an important step towards the clinical development of PRP, as we consider the possible sites to conduct a First-In-Human study in advanced cancer patients, possibly in Europe, specifically the UK, or at a prominent cancer hospital in Australia, with significant experience in early stage clinical development. Attractive R&D tax incentive benefits could be gained by undertaking the trial in Australia, as well as utilizing world-class facilities dedicated to treating and caring for people with cancer. We will investigate selected clinical trial sites more thoroughly as we commence preparation of a clinical trial application for PRP.
In March 2019, we presented at the 31st Annual ROTH Conference held at the Ritz Carlton, Laguna Niguel located in Orange County, CA. This conference featured presentations from public and private companies across a variety of industry sectors and is one of the largest of its kind in the US. Last year, the ROTH Conference hosted close to 550 participating companies and more than 4,700 attendees, including institutional investors, analysts, family offices and high net worth investors. As part of our presentation and one-on-one meetings, we provided a further Company update for 2019, including our focus on the advancement of our lead product, PRP.
In March 2019, we announced that we have received a Notice of Allowance from the United States Patent and Trademark Office (the “USPTO”) confirming composition of matter claims involving trypsinogen and chymotryosinogen have been allowed. The additional composition claims are a continuation from the original foundation patent in the U.S. and as a result, both method of treatment and composition claims will protect PRP, our lead product candidate. A Notice of Allowance is issued by the USPTO to indicate that it believes an invention qualifies for a patent. The reasons for allowance stipulated by the USPTO examiner stated that the scientific declarations presented establishes that compositions comprising trypsinogen and chymotrypsinogen exhibit a synergistic ability to inhibit the growth of various cancer cell lines, and that this effect would be unexpected to one of ordinary skill in the art, thus concluding the claims were patentable.
In February 2019, we presented at the 2019 BIO CEO & Investor Conference held at the New York Marriott Marquis. This conference is one of the largest investor conferences focused on established and emerging publicly traded and select private biotech companies. As part of our presentation and one-on-one meetings, we provided a Company update for 2019, including our focus on the advancement of our lead product, PRP, in 2019.
In January 2019, we announced that a cooperation agreement has been entered into between the University of Jaén and our Company to commence the POP1 joint drug discovery program to be co-funded by both parties. The agreement coincides with the appointment of research scientist, Mr. Aitor González, to lead the drug discovery and research activities over the next 3 to 4 years. The objective of the program is to identify and develop suitable backup compounds to our lead product candidate, PRP.As part of the agreement, Macarena Perán, Ph.D. and Julian Kenyon, M.D. have been appointed as joint supervisors, representing the University and our Company, respectively. The program involves advancing new compounds through a drug screening process, followed by preclinical and early stage clinical development. As the drug candidate progresses along the development pathway, the collaboration will also involve the Universities of Granada and Jaén, as well as Granada and Almeria Hospitals, which are members of FIBAO, a Public Health Foundation, based in Granada, Spain, committed to assisting commercial partners with the development and commercialization of innovative technologies designed to benefit humankind.
In December 2018, we announced that our foundation patent application has been granted by the Office of the Controller General of Patents, Design and Trademarks, India. The foundation patent, which covers our lead product candidate, PRP, pioneers the discovery of a pharmaceutical composition for treating cancer via a combination of trypsinogen and/or chymotrypsinogen pancreatic proenzymes. As of June 30, 2020, the foundation patent has been granted in the USA, Europe (including Belgium, Czech Republic, Denmark, France, Germany, Ireland, Italy, the Netherlands, Portugal, Spain, Sweden, Switzerland/Liechtenstein, Turkey and the United Kingdom), China, Japan, Indonesia, Malaysia, Israel, Australia, New Zealand, Singapore, South Africa, Mexico, Republic of Korea, Hong Kong and more recently, India. It is presently under examination in Canada and Brazil.
Financing Activities
In February 2019, we entered into the Equity Purchase Agreement (the “Equity Purchase Agreement”) with Oasis Capital, LLC, an institutional accredited investor (“Oasis Capital”) pursuant to which Oasis Capital committed to purchase up to $10,000,000 shares of our common stock (the “Equity Line”).
| 74 |
Effective July 3, 2019, the Company entered into a securities purchase agreement with Power Up Lending Group Ltd. (“Power Up”), pursuant to which Power Up purchased a convertible promissory note (the “July 2019 Power Up Note”) from the Company in the aggregate principal amount of $78,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Power Up. The transaction closed on July 3, 2019 and the Company received payment on July 8, 2019 in the amount of $78,000, of which $2,500 was paid directly toward legal fees and $500 to Power Up for due diligence fees resulting in net cash proceeds of $75,000.
Effective July 30, 2019, the Company entered into a securities purchase agreement with Odyssey Capital Funding LLC,. (“Odyssey”), pursuant to which Odyssey purchased a convertible promissory note (the “July 2019 Odyssey Note”) from the Company in the aggregate principal amount of $320,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Odyssey. The July 2019 Odyssey Note contains an original discount of $25,000. The transaction closed on July 30, 2019 and the Company received payment on August 1, 2019 in the amount of $295,000, of which $10,000 was paid directly toward legal fees, resulting in net cash proceeds of $285,000.
Effective August 30, 2019, the Company entered into a securities purchase agreement with Auctus Fund, LLC (“Auctus”), pursuant to which Auctus purchased a convertible promissory note (the “August 2019 Auctus Note”) from the Company in the aggregate principal amount of $550,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Auctus. The transaction closed on August 30, 2019 and the Company received payment on September 4, 2019 in the amount of $550,000, of which $5,000 was paid directly toward legal fees and $40,000 to Auctus for due diligence fees resulting in net cash proceeds of $505,000.
Effective October 1, 2019, the Company entered into a securities purchase agreement with GW Holdings, (“GW”), pursuant to which GW purchased a convertible promissory note (the “October 1, 2019 GW Note”) from the Company in the aggregate principal amount of $131,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of GW any time after the six month anniversary of October 1, 2019. The transaction closed on October 1, 2019 and the Company received payment on October 2, 2019 in the amount of $131,000, of which $6,000 was paid directly toward legal fees resulting in net cash proceeds of $125,000.
Effective October 3, 2019, the Company entered into a securities purchase agreement with Crown Bridge Partners, LLC (“Crown Bridge”), pursuant to which Crown Bridge purchased a convertible promissory note (the “October 3, 2019 Crown Bridge Note”) from the Company in the aggregate principal amount of $108,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Crown Bridge. The transaction closed on October 3, 2019 and the Company received payment on October 9, 2019 in the amount of $108,000, of which $3,000 was paid directly toward legal fees, and included an original issuance discount of $5,000 resulting in net cash proceeds of $100,000.
The Company entered into a Securities Purchase Agreement whereby an investor (the “First Investor”) purchased from the Company, for a purchase price of $75,000 (the “First Investor Purchase Price”) a Convertible Redeemable Promissory Note, in the principal amount of $75,000 (the “First Note”). The First Investor Purchase Price was funded to the Company on January 13, 2020. The First Note is due and payable on January 6, 2021 (the “First Investor Maturity Date”) and entitles the holder to 8% interest per annum (the “First Investor Interest Rate”). The First Note may be converted into shares of the Company’s common stock at any time during the period beginning on the date that is one hundred eighty (180) days following the date of issuance and ending on the later of (i) the First Investor Maturity Date and (ii) the date of payment of the default amount, as defined therein.
The Company entered into a Securities Purchase Agreement whereby a second investor (the “Second Investor”) purchased from the Company, for a purchase price of $105,000 (the “Second Purchase Price”) a Convertible Redeemable Promissory Note, in the principal amount of $110,250 (the “Second Note”). The Second Purchase Price was funded to the Company on January 13, 2020. The Second Note is due and payable on January 13, 2021 (the “Second Maturity Date”) and entitles the holder to 8% interest per annum (the “Interest Rate”). The Second Note may be converted into shares of the Company’s common stock at any time during the period beginning on the date of issuance and ending on the later of (i) the Second Maturity Date and (ii) the date of payment of the default amount, as defined therein.
| 75 |
The Company entered into a Securities Purchase Agreement whereby a third investor (the “Third Investor”) purchased from the Company, for a purchase price of $54,500 (the “Third Purchase Price”) a Convertible Redeemable Promissory Note, in the principal amount of $58,000 (the “Third Note”). The Third Purchase Price was funded to the Company on January 22, 2020. The Third Note is due and payable on January 22, 2021 (the “Third Maturity Date”) and entitles the holder to 10% interest per annum (the “Interest Rate”). The Third Note may be converted into shares of the Company’s common stock at any time during the period beginning on the date of issuance and ending on the later of (i) the Third Maturity Date and (ii) the date of payment of the default amount, as defined therein.
The Company entered into a Securities Purchase Agreement whereby a fourth investor (the “Fourth Investor”) purchased from the Company, for a purchase price of $75,000 (the “Fourth Purchase Price”) a Convertible Redeemable Promissory Note, in the principal amount of $75,000 (the “Fourth Note”). The Fourth Purchase Price was funded to the Company on March 4, 2020. The Fourth Note is due and payable on February 19, 2021 (the “Fourth Maturity Date”) and entitles the holder to 8% interest per annum. The Fourth Note may be converted into shares of the Company’s common stock at any time during the period beginning on the date of issuance and ending on the later of (i) the Fourth Maturity Date and (ii) the date of payment of the default amount, as defined therein.
The Company entered into a Securities Purchase Agreement whereby a fourth investor (the “Fifth Investor”) purchased from the Company, for a purchase price of $43,000 (the “Fifth Purchase Price”) a Convertible Redeemable Promissory Note, in the principal amount of $43,000 (the “Fifth Note”). The Fifth Purchase Price was funded to the Company on March 25, 2020. The Fifth Note is due and payable on March 12, 2021 (the “Fifth Maturity Date”) and entitles the holder to 8% interest per annum. The Fifth Note may be converted into shares of the Company’s common stock at any time during the period beginning on the date of issuance and ending on the later of (i) the Fourth Maturity Date and (ii) the date of payment of the default amount, as defined therein.
On April 3, 2020, the Company closed on a transaction related to a Securities Purchase Agreement (the “Securities Purchase Agreement”) entered into on March 30, 2020, whereby an investor (the “Investor”) purchased from the Company, 7,500,000 units (the “Units”), each consisting of (i) 1.5 shares of the Company’s common stock (the “Common Stock”), or pre-funded warrants (the “Prefunded Warrants” upon Investors election due to the 4.99% blocker provisions discussed below) and (ii) 1.5 warrants to purchase one share of Common Stock (“Series A Warrants”, and collectively with the Common Stock the “Units”). In addition to the Units, the Investor was issued 63,750,000 warrants to purchase one share of Common Stock (the “Series B Warrants”) and an additional 63,750,000 warrants to purchase one share of Common Stock, subject to a vesting schedule (the “Series C Warrants” and, together with the Prefunded Warrants, the Series A Warrants, and the Series B Warrants, the “Warrants”). The aggregate purchase price for the Units, the Series A Warrants, the Series B Warrants and the Series C Warrants of $450,000 was paid at closing (the “Purchase Price”) or $0.06 per unit purchase price. The Securities Purchase Agreement contains a blocker provision whereby the Investor or any of its affiliates would not beneficially own in excess of 4.99% of the outstanding number of shares of Common Stock (“Beneficial Ownership Limitation”). As such, the Investor may elect to purchase Prefunded Warrants equal to the same number of shares of Common Stock that the Company would have been issued.
Due to the Beneficial Ownership Limitation, the 11,250,000 shares of Common Stock underlying the Units issuable at closing of the Securities Purchase Agreement are comprised of 804,518 shares of restricted Common Stock and 10,445,482 Prefunded Warrants with exercise price of $0.0001 (but can be less than par value). The Prefunded Warrants shall be exercisable immediately and shall expire when exercised in full.
The Securities Purchase Agreement contains such representations, warranties and covenants as are typical for a transaction of this nature.
Series A Warrants
Pursuant to the Securities Purchase Agreement, the Investor purchased Series A Warrants to purchase up to 11,250,000 shares of Common Stock, subject to adjustment as provided therein. The Series A Warrants have a cash exercise price of $0.20 per share and are immediately exercisable and expire in 3 years. The Series A Warrants contain a provision for cashless exercise in the event there is no effective registration statement registering the shares underlying the Series A Warrants calculated based on the difference between the exercise price of the Series A Warrant and the trading price of the stock (the “Cashless Exercise”).Additionally, the Series A Warrants contain a provision for a cashless conversion at the Holder’s option should the trading price of the Common Stock fall below $0.20 per share calculated based on the difference between the exercise price of the Series A Warrant and 70% of the Market Price, as defined therein (the” Alternate Cashless Exercise”).
| 76 |
Series B Warrants
Pursuant to the Securities Purchase Agreement, the Investor purchased Series B Warrants to purchase up to 63,750,000 shares of Common Stock, subject to adjustment as provided therein; provided, however, commencing on the 90th day following the effective date, the Company may reduce the number of Warrant Shares issuable upon exercise thereof by 37,500,000 upon 10 Trading Days’ prior written notice to the Holder provided that the Company issues to the Holder 3,750,000 shares of Common Stock (or, at the election of the Holder, an equivalent number of pre-funded warrants) and Series A Warrants to purchase up to 3,750,000 shares of Common Stock, which shares shall be issued pursuant to a registration statement without restrictions on resale. The Series B Warrants have a cash exercise price of $0.04 per share. The Series B Warrants contain a provision for Cashless Exercise.
Series C Warrants
Pursuant to the Securities Purchase Agreement, the Investor purchased Series C Warrants to purchase up to 63,750,000 shares of Common Stock, subject to adjustment as provided therein and expire in 3 years. The Series C Warrants have a cash exercise price of $0.20 per share, subject to a vesting schedule, which is based on such Holder’s exercise of the Series B Warrants. The Series C Warrants contain provisions for Cashless Exercise and Alternate Cashless Exercise.
Critical Accounting Estimates
Below is a discussion of our more subjective accounting estimation processes for purposes of explaining (i) the methodology used in calculating the estimates, (ii) the inherent uncertainties pertaining to such estimates, and (iii) the possible effects of a significant variance in actual experience, from that of the estimate, on our financial condition. Estimates involve numerous assumptions that, if incorrect, could create a material adverse impact on the Company’s results of operations and financial condition.
Reference is frequently made herein to the Financial Accounting Standards Board (the “FASB”) Accounting Standards Codification (“ASC”). This is the source of authoritative US GAAP recognized by the FASB to be applied to non-governmental entities. Each ASC reference in this filing is presented with a three-digit number, which represents its Topic. As necessary for explanation and as applicable, an ASC topic may be followed with a two-digit subtopic, a two-digit section or a two-or-three digit paragraph.
Foreign Currency Translation and Comprehensive Income (Loss): The Company’s wholly owned subsidiary’s functional currency is the AUD. For financial reporting purposes, the Australian Dollar (“AUD”) has been translated into USD as the Company’s reporting currency. Assets and liabilities are translated at the exchange rate in effect at the balance sheet date. Revenues and expenses are translated at the average rate of exchange prevailing during the reporting period. Equity transactions are translated at each historical transaction date spot rate. Translation adjustments arising from the use of different exchange rates from period to period are included as a component of stockholders’ equity (deficit) as “accumulated other comprehensive income (loss).” Gains and losses resulting from foreign currency transactions are included in the statement of operations and comprehensive loss as other income (expense).
Accounting for Income Taxes: We are governed by Australian and United States income tax laws, which are administered by the Australian Taxation Office and the United States Internal Revenue Service, respectively. We follow ASC 740, “Accounting for Income Taxes,” which requires an asset and liability approach to financial accounting and reporting for income taxes. Deferred income tax assets and liabilities are computed annually for temporary differences between the financial statements and tax bases of assets and liabilities that will result in taxable or deductible amounts in the future based on enacted tax laws and rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized. Income tax expense is the tax payable or refundable for the period plus or minus the change during the period in deferred tax assets and liabilities.
The Company adopted provisions of ASC 740, Sections 25 through 60, “Accounting for Uncertainty in Income Taxes.” These sections provide detailed guidance for the financial statement recognition, measurement and disclosure of uncertain tax positions recognized in the financial statements. Tax positions must meet a “more-likely-than-not” recognition threshold at the effective date to be recognized upon the adoption of ASC 740 and in subsequent periods.
Accounting for Stock Based Compensation: We record stock based compensation in accordance with ASC 718, “Stock Compensation” and Staff Accounting Bulletin No. 107 issued by the SEC in March 2005 regarding its interpretation of ASC 718. ASC 718 requires the fair value of all stock-based employee compensation awarded to employees to be recorded as an expense over the related requisite service period. The statement also requires the recognition of compensation expense for the fair value of any unvested stock option awards outstanding at the date of adoption. We value any employee or non-employee stock based compensation at fair value using the Black-Scholes Option Pricing Model.
| 77 |
We account for non-employee share-based awards in accordance with the measurement and recognition criteria of ASC 718.
Derivative Instruments: ASC 815, “Derivatives and Hedging,” establishes accounting and reporting standards for derivative instruments and for hedging activities by requiring that all derivatives be recognized in the balance sheet and measured at fair value. Gains or losses resulting from changes in the fair value of derivatives are recognized in earnings. On the date of conversion, or payoff, of debt, we record the fair value of the conversion shares, remove the fair value of the related derivative liability, remove any discounts and record a net gain or loss on debt extinguishment.
Convertible Notes with Variable Conversion Options: We have entered into convertible notes, some of which contain variable conversion options, whereby the outstanding principal and accrued interest may be converted, by the holder, into common shares at or around a fixed discount to the price of the common stock at the time of conversion. We treat these convertible notes as stock settled debt under ASC 480 and measure the fair value of the notes at the time of issuance, which is the result of the share price discount at the time of conversion, and records the put premium as accretion to interest expense.
Research and Development Tax Credits: We may apply for Research and Development tax concessions with the Australian Taxation Office on an annual basis. Although the amount is possible to estimate at year end, the Australian Taxation Office may reject or materially alter the claim amount. Accordingly, we do not recognize the benefit of the claim amount until cash receipt since collectability is not certain until such time. The tax concession is a refundable credit. If we have net income then we can receive the credit which reduces its income tax liability. If we have net losses, then we may still receive a cash payment for the credit, however, our net operating loss carry forwards are reduced by the gross equivalent loss that would produce the credit amount when the income tax rate is applied to that gross amount. The concession is recognized as an income tax benefit, in operations, upon receipt.
Recent Accounting Pronouncements
Please see section captioned “Recent Accounting Pronouncements” in Note 1 to our consolidated financial statements included in the registration statement of which this prospectus forms a part for a discussion of recently issued and adopted accounting pronouncements.
Reverse Stock Split Presentation
The share and per share figures in this Management’s Discussion and Analysis of Financial Condition and Results of operations section, along with elsewhere in this annual report, have been adjusted to reflect the 1-for-500 Reverse Stock Split of our authorized and outstanding shares of common stock, which occurred on June 24, 2019.
Results of Operations
The following discussion of our results of operations for the periods presented should be read in conjunction with our consolidated financial statements and notes thereto included elsewhere in this prospectus. The results discussed below are of our Company and our wholly-owned Australian subsidiary, Propanc PTY LTD.
Fiscal Year Ended June 30, 2020, as compared to the Fiscal Year Ended June 30, 2019
Revenue
For the fiscal years 2020 and 2019 we generated no revenue because we are currently undertaking research and development activities for market approval and no sales were generated in this period.
Administration Expense
Administration expense increased to $3,281,464 for the year ended June 30, 2020 as compared to $2,326,350 for the year ended June 30, 2019. This increase of approximately $955,000 is primarily attributable to an increase of approximately $1,154,000 in stock-based expenses for services, an increase of approximately $119,000 in capital raising costs, an increase in general consulting and accountancy fee of approximately $64,000, an increase in insurance expense of approximately $17,000, an increase of approximately $139,000 in marketing and market research expense, increase of approximately $3,000 of other general and administrative expenses and, offset by decrease of approximately $122,000 in employee remuneration expense as a result of a one off adjustment in employee leave liability of approximately $157,000 in the year ended June 30, 2019, decreases in investor relations based expense of approximately $80,000, a decrease of approximately $69,000 in general legal and intellectual property legal expense, a decrease of approximately $42,000 in public company filing fees, a decrease of $45,000 in travel expense and a decrease of approximately $183,000 in employee leave accruals which is a result of an adjustment of approximately $176,000 to employee leave accruals during the year ended June 30, 2019 in order to comply with Australian leave entitlements.
| 78 |
Occupancy Expense
Occupancy expense increased by approximately $4,700 to $32,809 for the year ended June 30, 2020. The increase primarily relates to rent adjustment on under paid rent of $4,800 during the year ended June 30, 2020.
Research and Development Expenses
Research and development expenses were $179,987 for the year ended June 30, 2020, as compared to $260,335 for the year ended June 30, 2019. The decrease in research and development expenses is primarily attributable to completion of process development activities and preparation for commencement of the engineering run and subsequent full scale GMP manufacture of PRP for clinical trials, with the process, preparation and small scale manufacture having been completed in the period ended December 31, 2017, which the clinical trials we hope to commence in 2021 calendar year, if we raise sufficient proceeds by raising additional capital. Completed activities include raw material purification and stabilization process development, development of analytical quality assurance and control methods, reproduction runs for raw materials, and preparation of raw materials and finished product specifications for future full scale GMP manufacture of PRP.
Interest Expense/Income
Interest expense increased to $1,748,381 for the year ended June 30, 2020, as compared to $1,314,539 for the year ended June 30, 2019. Interest expense is primarily comprised of approximately $734,130 of debt discount amortization and approximately $836,724 accretion of debt premium. This increase is primarily attributable to an increase in the issuance of debt containing embedded derivatives resulting in an increase amortization of debt discount, along with the increase in issuance of convertible notes with discounted debt features during the year ended June 30, 2020.
Change in Fair Value of Derivative Liabilities
Change in fair value of derivative liabilities changed by $2,857,439, to a gain of $385,293 for the year ended June 30, 2020, as compared to a loss of $(2,472,146) for the year ended June 30, 2019. This change is primarily attributable to an increase in the volatility of the prices of our shares of common stock along with a decrease in stock price during the year ended June 30, 2020, which resulted in the recognition of a smaller loss from change in fair value.
Gain on Debt Settlements, Net
There were no debt settlements during the year ended June 30, 2020 and as a result there were no gains or (losses) on settlement of debt during that period, as compared with a gain of $14,101 for the year ended June 30, 2019.
Gain (loss) on Extinguishment of Debt, net
During the year ended June 30, 2020, notes with principal amounts of $254,500 and accrued interest of $15,408 contained bifurcated embedded conversion option derivatives. Accordingly, the fair market value of the shares issued was $565,746 resulting in a loss on extinguishment at the time of conversion of $295,838 and $362,961 of derivative fair value was recorded as a gain on extinguishment at the time of conversion. During the year ended June 30, 2019, the Company repaid three convertible notes which were treated as derivative instruments, incurred penalties of $92,133 and recorded a gain on the removal of the derivatives of $936,650. Additionally, the Company issued shares of common stock with a value of $1,335,047 which resulted in a gain on extinguishment of $359,725 as the note and derivative fair value exceeded the fair value of shares converted.
Foreign Currency Transaction Gain (Loss)
Foreign currency transaction decreased to a loss of $(143,808) for the year ended June 30, 2020 as compared with a loss of $(690,748) for the year ended June 30, 2019. The decrease in foreign currency transaction loss is primarily attributable to greater fluctuation in exchange rates during the year ended June 30, 2020, as compared to during the year ended June 30, 2019, and 2020 having less intercompany loans.
Income Tax Benefit
During the years ended June 30, 2020 and 2019, the Company applied for and received from the Australian Taxation Office a research and development tax credit in the amount of $134,728 and $115,437, respectively.
| 79 |
Net loss
Net loss decreased to $(4,740,723) for the year ended June 30, 2020 as compared to a net loss of $(5,758,369) for the year ended June 30, 2019. The change relates to factors discussed above.
Liquidity and Capital Resources
Current Financial Condition
As of June 30, 2020, we had total assets of $98,518, comprised primarily of cash of $67,007, GST tax receivable of $2,015, property and equipment, net, of $5,747 and operating lease right of use asset, net, $21,682. This compares with total assets of $101,652 as of June 30, 2019, comprised primarily of cash of $2,394, GST tax receivable of $5,439, prepaid expenses and other current assets of $83,299 and property and equipment, net, of $8,417.
We had current liabilities of $3,739,943, primarily comprised of net convertible debt of $1,557,734, accounts payable and accrued expenses of $1,544,387 and embedded conversion option liabilities of $354,109, as of June 30, 2020. This compares with current liabilities of $4,402,888, primarily comprised of net convertible debt of $1,657,377, accounts payable and accrued expenses of $1,640,379 and embedded conversion option liabilities of $698,264, as of June 30, 2019.
We have funded our operations primarily through the issuance of equity and/or convertible securities for cash. The cash was used primarily for payments for research and development, administration expenses, occupancy expenses, professional fees, consultants and travel.
During the year ended June 30, 2020, we borrowed amounts of approximately $1,591,250 from the sale of convertible promissory notes during such period with various maturity dates ranging from July 3, 2020 to March 12, 2021.
We have substantial capital resource requirements and have incurred significant losses since inception. As of June 30, 2020, we had $67,007 in cash. We depend upon debt and/or equity financing to fund our ongoing operations and to execute our current business plan. Such capital requirements are in excess of what we have in available cash and for which we currently have commitments. Therefore, we presently do not have enough available cash to meet our obligations over the next 12 months. If continued funding and capital resources are unavailable at reasonable terms, we may curtail our plan of operations. We will be required to obtain alternative or additional financing from financial institutions, investors or otherwise, in order to maintain and expand our existing operations. The failure by us to obtain such financing would have a material adverse effect upon our business, financial condition and results of operations, and adversely affecting our ability to complete ongoing activities in connection with our research and development programs.
Sources and Uses of Cash
For
the Fiscal Year June 30, |
||||||||
| 2020 | 2019 | |||||||
| Net cash used in operating activities | $ | (1,849,589 | ) | $ | (2,060,037 | ) | ||
| Net cash used in investing activities | $ | - | $ | (2,874 | ) | |||
| Net cash provided by financing activities | $ | 1,890,240 | $ | 2,118,560 | ||||
| Effect of exchange rate changes on cash | $ | 23,962 | $ | (73,176 | ) | |||
| 80 |
Net cash used in operating activities was $1,849,589 for the fiscal year ended June 30, 2020 compared to $2,060,037 for the fiscal year ended June 30, 2019. This decrease is primarily due to a decrease in change in fair value of derivative liabilities of approximately $2,857,000, decrease in accrued expenses of approximately $21,000, decrease in employment related liabilities of approximately $153,000, changes in foreign currency transaction loss of approximately $547,000 offset by increases primarily due to increase in stock-based compensation of $1,154,000, amortization of debt discount of $344,000, accretion of put premium of $131,000, increase in gain on extinguishment of debt of approximately $1,229,000, increase in prepaid expenses and other assets of $166,000 and accounts payable of $105,000.
Cash flows provided by financing activities for the fiscal year ended June 30, 2020 were $1,890,240 as compared to $2,118,560 for the fiscal year ended June 30, 2019. During the year ended June 30, 2020, we received net proceeds from the issuance of convertible promissory notes of $1,465,250 and net proceeds from sale of common stock of $424,990. During the year ended June 30, 2019, we received net proceeds from the issuance of convertible promissory notes of $1,305,150 and sale of common stock of $1,085,380 net, offset by repayments of convertible notes of $272,000.
The effect of the exchange rate on cash resulted in a $23,962 positive adjustment to cash flows in the year ended June 30, 2020 as compared to a negative adjustment of $73,176 to cash flows in the year ended June 30, 2019. The reason for the fluctuation is due to the application of currency translation rates throughout the cash flow statement, the volume of transactions within each period and the daily fluctuation in exchange rates.
Going Concern Qualification
We did not generate any revenue for the fiscal years ended June 30, 2020 and 2019 and have incurred significant losses and cash used in operations, and such losses and use of cash are expected to continue. Our independent registered public accounting firm has included a “Going Concern Qualification” in their audit report for each of the fiscal years ended June 30, 2020 and 2019. In addition, we have negative working capital and convertible debt that is past maturity that we are currently negotiating with lenders in order to amend the maturity dates. The foregoing raises substantial doubt about our ability to continue as a going concern for a period of 12 months from the issuance date of the consolidated financial statements. Our ability to continue as a going concern is dependent on our ability to execute our strategy and on our ability to raise additional funds and/or to consummate a public offering. Management is currently seeking additional funds, primarily through the issuance of equity and/or debt securities for cash to operate our business. No assurance can be given that any future financing will be available or, if available, that it will be on terms that are satisfactory to us. Even if we are able to obtain additional financing, it may contain undue restrictions on our operations, in the case of debt financing or cause substantial dilution for our stockholders, in case of equity and/or convertible debt financing. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty. The “Going Concern Qualification” might make it substantially more difficult to raise capital.
| 81 |
DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The following table sets forth certain information regarding our current executive officers and directors as of October 13, 2020:
| Name | Age | Position | |||
| James Nathanielsz | 46 | Chief Executive Officer, Chairman, Secretary, Treasurer and Director | |||
| Dr. Julian Kenyon | 73 | Chief Scientific Officer and Director | |||
| Carlo Campiciano | 66 | Chief Financial Officer and Secretary |
Executive Officers and Directors
James Nathanielsz ― Mr. Nathanielsz has served as a director of our Company since inception. Mr. Nathanielsz has served as a director and chief executive officer of our Australian subsidiary since October 2007. From July 2006 until October 2007, Mr. Nathanielsz served as the New Products Manager of Biota Holdings Limited, an anti-infective drug development company in Australia. Mr. Nathanielsz graduated with a Bachelor of Applied Science, majoring in Biochemistry/Applied Chemistry and with a Master of Entrepreneurship & Innovation from Swinburne University of Technology in Melbourne, Australia.
| 82 |
Our board of directors has concluded that Mr. Nathanielsz is well-qualified to serve on our board of directors and has the requisite qualifications, skills and perspectives based on, among other factors, him being a Co-Founder of our Australian company and for his experience in research and development and manufacturing and distribution, as well as him being our controlling stockholder, and his significant business, investment, finance and public company experience, particularly with biotech companies.
Dr. Julian Kenyon ― Dr. Kenyon has served as our Chief Scientific Officer since inception (being appointed as an executive officer on May 14, 2019) and a director of our Company and since inception. Dr. Kenyon co-founded our Australian subsidiary and was appointed as a director of our Australian subsidiary on February 12, 2008. Since 2000, Dr. Kenyon has served as an integrated medical physician and Medical Director of the Dove Clinic for Integrated Medicine in Winchester and London. Dr. Kenyon graduated from the University of Liverpool with a Bachelor of Medicine and Surgery and with a research degree, Doctor of Medicine. Since 1972, he was appointed a Primary Fellow of the Royal College of Surgeons, Edinburgh.
Our board of directors has concluded that Dr. Kenyon is well-qualified to serve on our board of directors and has the requisite qualifications, skills and perspectives based on, among other factors, him being a Co-Founder of our Australian subsidiary and because our business is based on his initial work at the Dove Clinic.
Carlo Campiciano ― Mr. Campiciano is qualified as a chartered accountant and has extensive experience working with business on a wide range of areas including taxation, finance, operations, planning, operational and financial strategy. Prior to his appointment as the Company’s Chief Financial Officer and Secretary, Mr. Campiciano served as the Chief Financial Officer of MedAdvisor International Pty Ltd. from July 2012 until December 2016, where he was part of the foundation team that launched MedAdvisor in 2012 and since its launch has been key in raising several rounds of capital as well the company’s listing on the ASX in December 2015. As part of MedAdvisor’s executive team, Mr. Campiciano was instrumental in the strategic and operational development of the company’s business, which included overseeing the expansion of its operations to be a market leader in Australia, as well as establishing the business in the USA, UK and Asia. Mr. Campiciano also served as Chief Financial Officer for TGA Unlimited Pty Ltd., a start-up business which he helped grow from annual revenues of $2 million to over $40 million. Mr. Campiciano has also run his own private accounting practice for over 30 years and has spent 12 years lecturing in venture finance in the Master of Entrepreneurship and Innovation program at the Swinburne Graduate School of Entrepreneurship (Melbourne, Australia). Mr. Campiciano commenced his career at Coopers & Lybrand (currently PricewaterhouseCoopers). Mr. Campiciano has a Masters of Entrepreneurship and Innovation from Swinburne University of Technology (Melbourne, Australia), Graduate Diploma in Computing from Monash University Caulfield (formerly Caulfield Institute of Technology) (Melbourne, Australia) and Bachelor of Business (Accounting) from RMIT University (Melbourne, Australia) and Certificate in Corporate Governance from the Governance Institute of Australia. Mr. Campiciano is a member of the Institute of Public Accountants of Australia and has received his US GAAP certification.
Our board of directors has concluded that Mr. Campiciano is well-qualified to serve on our board of directors and has the requisite qualifications, skills and perspectives based on, among other factors, his extensive experience as a Chief Financial Officer and as a chartered accountant.
Key Consultants
Professor Klaus Kutz ― Professor Kutz has been serving as our acting Chief Medical Officer in a consulting capacity since December 2010. Subject to the completion of this offering and as our resources allow, we hope to appoint Professor Kutz to serve as our Chief Medical Officer in a more active executive capacity in the second half of 2019 calendar year. Professor Kutz has over 20 years of experience as an independent consultant in Clinical Pharmacology and Safety for pharmaceutical companies and clinical research organizations. His specialty over the last six years is Oncology, including preparation of multiple NDAs and INDs for small and medium sized pharmaceutical companies. He has prepared, organized and reported clinical Phase I studies in oncology and Phase II studies in different cancer indications (prostate, gastric, ovarian, small cell lung cancer) and Non-Hodgkin Lymphomas. Professor Kutz has more than 13 years of experience as Head of Clinical Pharmacology with world-wide responsibilities for Phase I and Clinical Pharmacokinetics in two internationally operating pharmaceutical companies, setting up and restructuring international Clinical Pharmacology departments. His achievements include the successful world-wide registration of multiple important Sandoz’ compounds by preparation of multiple NDAs (New Drug Applications) and Expert reports (including Written Summary), as well as the preparation of multiple INDs (Investigational New Drug Applications) for Sandoz Pharma Ltd and Sanofi Research. Professor Kutz is a specialist for Internal Medicine, Gastroenterology, and Clinical Pharmacology and he is also Professor of Medicine at the University of Bonn, Germany.
Term of Office
Our directors are appointed for a one-year term to hold office until the next annual general meeting of our stockholders or until removed from office in accordance with our Bylaws and the provisions of the Delaware General Corporation Law. Our directors hold office after the expiration of his or her term until his or her successor is elected and qualified, or until his or her resignation, death or removal in accordance with our Bylaws or the Delaware General Corporation Law.
Our officers are appointed by our board of directors and hold office until removed by our board of directors at any time for any reason.
Family Relationships
There are no family relationships between or among any of our directors or executive officers or persons nominated or chosen by us to become directors or executive officers.
| 83 |
Director Independence
Our board of directors has reviewed the independence of our directors and has determined that none of our directors qualifies as an independent director pursuant to Rule 5605(a)(2) of Nasdaq and applicable SEC rules and regulations. In making this determination, our board of directors considered the relationships that each of our directors has with us and all other facts and circumstances our board of directors deemed relevant in determining their independence.
Board Committees
Our board of directors has no separately designated committees and our two-member board of directors carries out the functions of both an audit committee and a compensation committee. We do not have an audit committee financial expert serving on our board of directors. Due to our limited financial resources, we are not in a position to retain an independent director with the qualifications to serve as an audit committee financial expert at this time.
Scientific Advisory Board
We have a Scientific Advisory Board that provides advice to our management relating to the following:
| ● | The identification, assessment, evaluation, selection, conduct and management of research projects, both those which are under review and are in progress; | |
| ● | Intellectual property; and | |
| ● | Commercialization. |
The Scientific Advisory Board may also address issues related to improving project selection, formal review processes and management procedures within our Company. The Scientific Advisory Board will generally be composed of an advisory panel of clinicians with expertise in translational research.
As of October 13, 2020, the members of the Scientific Advisory Board were:
| ● | Professor John Smyth; | |
| ● | Professor Klaus Kutz (also serving as our acting Chief Medical Officer); | |
| ● | Dr. Joseph Chalil; | |
| ● | Dr. Macarena Perán; | |
| ● | Dr. Juan Antonio Marchal Corrales; | |
| ● | Dr. Maria Garcia; and | |
| ● | Dr. Ralf Brandt. |
Each of the members of our Scientific Advisory Board acts as an independent consultant and is compensated on an hourly basis for his or her services. There is presently no stock based compensation for their services. In addition, we may have relationships with entities with which the members may be associated.
Professor Kutz is also acting as Chief Medical Officer for Propanc in a non-executive capacity. His compensation continues to be based on an hourly rate as per his Advisory Board Agreement. Propanc intends to appoint Professor Kutz as Chief Medical Officer of Propanc in a full-time executive officer capacity at a time that is mutually agreed upon between both parties.
| 84 |
Professor John Smyth ― John Smyth has, for over 25 years, served as Chair of Medical Oncology in the University of Edinburgh Medical School, where his major research interest is the development and evaluation of new anti-cancer drugs. He has published over 300 papers and is Editor-in-Chief of the European Journal of Cancer. He served for several years on the UK Committee on Safety of Medicines, currently Chair’s the Expert Advisory Group for Oncology & Hematology for the Commission on Human Medicines and serves on the Expert Oncology Advisory Group to the European Drug Licensing Board. He is a fellow of the Royal College of Physicians of Edinburgh and London, and fellow of the Royal Society of Edinburgh. He is a past-president of the European Society of Medical Oncology and from 2005 to 2007 was President of the Federation of European Cancer Societies.
Professor Klaus Kutz ― Professor Kutz has over 20 years of experience as an independent consultant in Clinical Pharmacology and Safety for pharmaceutical companies and clinical research organizations. His specialty over the last six years is Oncology, including preparation of multiple NDAs and INDs for small and medium sized pharmaceutical companies. He has prepared, organized and reported clinical Phase I studies in oncology and Phase II studies in different cancer indications (prostate, gastric, ovarian, small cell lung cancer) and Non-Hodgkin Lymphomas. Professor Kutz has more than 13 years of experience as Head of Clinical Pharmacology with world-wide responsibilities for Phase I and Clinical Pharmacokinetics in two internationally operating pharmaceutical companies, setting up and restructuring international Clinical Pharmacology departments. His achievements include the successful world-wide registration of multiple important Sandoz’ compounds by preparation of multiple NDAs (New Drug Applications) and Expert reports (including Written Summary), as well as the preparation of multiple INDs (Investigational New Drug Applications) for Sandoz Pharma Ltd and Sanofi Research. He is a specialist for Internal Medicine, Gastroenterology, and Clinical Pharmacology and he is also Professor of Medicine at the University of Bonn, Germany.
Dr. Joseph Chalil ― Dr. Chalil is a Physician and Executive at Boehringer Ingelheim, the world’s largest privately held pharmaceutical company. Headquartered in Ingelheim, Germany, Boehringer Ingelheim operates globally with 146 affiliates and a more than 47,700 employees. In 2014, Boehringer Ingelheim achieved net sales of about 13.3 billion Euros. Research and development expenditure corresponds to 19.9 percent of its net sales. In addition to his responsibilities at Boehringer Ingelheim, Dr. Chalil is the Chairman of Global Clinical Research and Trial Network of the American Association of Physicians of Indian Origin (AAPI) and has served as Scientific Advisor to AAPI for the past five years. AAPI is the second largest physician organization in the U.S. second only to AMA, and the largest ethnic medical organization in the country. A veteran of the United States Navy Medical Corps, Dr. Chalil is also board certified in healthcare management, and has been awarded Fellowship by the American College of Healthcare Executives, an international professional society of more than 40,000 healthcare executives who lead hospitals, healthcare systems and other healthcare organizations. Dr. Chalil is an expert in U.S. Healthcare policy and a strong advocate for patient centered care and has also served as an advisor to various national political campaigns on healthcare issues. Dr. Chalil completed his higher studies in University of Medicine and Dentistry of New Jersey, Davenport University, JJM Medical College and Baylor College of Medicine. He has been a Visiting Professor at various Universities and serves on various company Boards.
Dr. Macarena Perán ― Dr. Macarena Perán holds a B.S. in Biology and an M.S. in Biochemistry and Molecular Biology from the University of Málaga, Spain. Dr. Perán moved to the Neuroscience Department at Durham University, UK, where she studied the Cellular Distribution and Immobilisation of GABAA Receptors on the cell membrane and graduated in 2000 with a Ph.D. She moved back to Spain and completed another Ph.D. program in the Faculty of Medicine focused on Changes in the Behavior of Central Nervous Proteins; she completed a second Ph.D. from Granada University. In 2005/2006, she attended Bath University, UK, Prof. David Tosh lab, and changed her research interest to the development of new anti-cancer drugs and cell therapy for regenerative medicine. In 2011, she spent a year as a visiting scientist in the Salk Institute for Biological Studies, California, Prof. Juan Carlos Izpisua-Belmonte lab. Currently, Dr. Perán is Reader in Anatomy at University of Jaén in Spain and is working with the Institute for Regenerative Medicine and Pathobiology (IBIMER).
Dr. Juan Antonio Marchal Corrales ― Dr. Juan Antonio Marchal Corrales is Professor of Anatomy and Embryology at the Faculty of Medicine of University of Granada. He graduated in Medicine and Surgery in 1992, obtaining the degree “summa cum laude”. He defended his doctoral thesis in 1996. Prof. Marchal has worked at three universities in different educational categories and is responsible for the research group “Differentiation, Regeneration and Cancer”. He has participated in 39 research projects of national and international character, being principal investigator in 13 of them. He has a total of 145 publications in journals, of which 125 are listed in the Journal Citation Reports. He has spent time at the University of Sassari (Italy) and as visiting professor. He is inventor of 14 patents, 4 of them licensed. He is a member of the Advisory Board of the International Graduate School of the University of Granada, member of the standing committee of the Scientific Council and coordinator of Area Research in the Biosanitary Institute of Granada (ibs.GRANADA) and member of the Governing Board at the Institute of Pathobiology and Regenerative Medicine (IBIMER). He has recently been named director of the Chair Drs. Galera and Requena of Cancer Stem Cell Research at the University of Granada.
| 85 |
Dr. Maria Garcia ― Dr. Maria Garcia, graduated in Biology from University of Granada (Spain) in 1997, became a Molecular Biologist working in the National Centre of Biotechnology characterizing the mechanism of action of “Protein kinase induced by interferon: PKR”. These studies gave rise to a PhD title awarded with an Extraordinary Thesis Award by the Autonomous University of Madrid in 2004. In 2002, Dr. García completed a 3-months stay at the University of Wyoming with Dr. Roth. During the postdoctoral period, she got major public and private funding to characterize new activity of the main tumor suppressor genes that are mutated in more than 50% of human cancers such as p53, ARF and Rb. Dr. García currently has a competitive research contract from the National Health System to lead translational cancer research, aiming at the integration of basic, clinical and epidemiological cancer research in the University Hospital Complex of Granada. She leads a line of research involving new antitumor drugs, biological therapies, biomarkers and cancer stem cell studies. Finally, Dr. García has more than 30 peer-reviewed publications in international journals with an average impact factor of 5 and a H-Index of 14.
Dr. Ralf Brandt ― Dr. Brandt is the co-founder of vivoPharm, a global oncology and immuno-oncology discovery services company providing a range of preclinical services, which merged and became a part of Cancer Genetics, Inc., a Nasdaq listed company enabling precision medicine in oncology from bench to bedside. Dr. Brandt currently serves as President of Discovery and Early Development of Cancer Genetics.Dr. Brandt is a biochemist and cell biologist with over 15 years experience in research programs of experimental oncology. He has immense experience in in vivo pharmacology and anti-cancer drug profiling. Dr. Brandt received his Licence (BSc in Biochemistry and Animal Physiology) in 1986, and his PhD (in Biochemistry) in 1991 from the Martin-Luther University of Halle-Wittenberg, Germany. Dr. Brandt was employed at research positions at the National Cancer Institute in Bethesda, MD, USA and at Schering AG, Germany. Since 1990, Dr. Brandt has been active in the field of preclinical oncology. He led the Tumour Biology program at Novartis Pharma AG, Switzerland and established several transgenic mouse lines developing tumours under the control of oncogenes. During Dr. Brandt’s long career in the pharmaceutical industry he has acquired significant knowledge and expertise in leading business units and representation of services to the pre-clinical research market. Dr. Brandt is also a member of the Scientific Advisory Board at Receptor Inc. in Toronto Canada.
Board Leadership Structure
Currently, the office of Chairman of our board of directors and Chief Executive Officer are held by Mr. Nathanielsz. Due to our size and early stage of operations, we believe it is currently most effective to have the Chairman of the board of directors and Chief Executive Officer positions be held by the same individual.
Risk Oversight
Our board of directors will oversee a company-wide approach to risk management. Our board of directors will determine the appropriate risk level for us generally, assess the specific risks faced by us and review the steps taken by management to manage those risks. While our board of directors will have ultimate oversight responsibility for the risk management process, its committees will oversee risk in certain specified areas.
Until we have established our compensation committee of our board of directors, our board of directors will be responsible for overseeing the management of risks relating to our executive compensation plans and arrangements, and the incentives created by the compensation awards it administers. Until we have established our audit committee, our board of directors will oversee management of enterprise risks and financial risks, as well as potential conflicts of interests. Our board of directors will be responsible for overseeing the management of risks associated with the independence of our board of directors.
| 86 |
Code of Ethics
The Board has adopted a Code of Ethics (the “Code”) to apply to all of our directors, officers and employees. The Code is intended to promote ethical conduct and compliance with laws and regulations, to provide guidance with respect to the handling of ethical issues, to implement mechanisms to report unethical conduct, to foster a culture of honesty and accountability, to deter wrongdoing and to ensure fair and accurate financial reporting. A copy of the Code is available at our website www.propanc.com.
Compensation Committee Interlocks and Insider Participation
None of our executive officers currently serves, or in the past three years has served, as a member of the board of directors or compensation committee of another entity that has one or more executive officers serving on our board of directors or the compensation committee. No member of our compensation committee has any other business relationship or affiliation with us other than his or her service as a director.
Section 16(A) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our directors, executive officers, and persons who own more than 10% of our common stock to file initial reports of ownership and changes in ownership of our common stock and other equity securities with the SEC. These individuals are required by the regulations of the SEC to furnish us with copies of all Section 16(a) forms they file. Based solely on a review of the copies of the forms furnished to us, and written representations from reporting persons that no Forms 5 were required to report delinquent filings, we believe that all filing requirements applicable to our officers, directors and 10% beneficial owners were complied with during the fiscal year ended June 30, 2019.
Nominations to the Board of Directors
General — Our directors take a critical role in guiding our strategic direction and oversee the management of the Company. Our board of directors’ candidates are considered based upon various criteria, such as their broad-based business and professional skills and experiences, a global business and social perspective, concern for the long-term interests of the stockholders, diversity, and personal integrity and judgment. In addition, directors must have time available to devote to our board of directors activities and to enhance their knowledge of our business. Accordingly, we seek to attract and retain highly qualified directors who have sufficient time to attend to their substantial duties and responsibilities to our Company.
Changes to the Procedures by Which Security Holders May Recommend Nominees to Our Board of Directors — During the year ended June 30, 2019, there were no material changes to the procedures by which our security holders may recommend nominees to our board of directors.
For the fiscal years ended June 30, 2019 and 2018, our sole named executive officer, who is our principal executive officer (the “Named Executive Officer”), was Mr. James Nathanielsz, our current Chief Executive Officer, Chairman and a director.
Summary Compensation Table
The following table sets forth the compensation paid or accrued by us to our Named Executive Officer for the fiscal years ended June 30, 2020 and 2019.
The compensation reported in the summary compensation table below is not necessarily indicative of how we will compensate our sole executive officer in the future. We expect that we will continue to review, evaluate and modify our compensation framework and the compensation of our officer could change as the business develops.
| Year | Salary ($) | Bonus ($) | Option Awards ($) | All
Other Compensation ($) | Total
($) | |||||||||||||||||||
| James Nathanielsz(1) | 2020 | $ | 357,988 | (2) | $ | 165,384 | (3) | - | $ | 66,821 | (4) | $ | 590,193 | |||||||||||
| Chief Executive Officer (June 30, 2020) | 2019 | $ | 421,916 | (2) | $ | 322,414 | (3) | $ | 165,747 | $ | 78,889 | (4) | $ | 988,966 | ||||||||||
| (1) | For purposes of the information included in the table, the conversion rates as of June 30, 2020 and 2019, $0.6891 and $0.7153, respectively, were used to convert amounts from AUD to USD. | |
| (2) | Under the Nathanielsz Employment Agreement (as defined below), Mr. Nathanielsz received a gross annual salary of $400,000 AUD per year effective February 1, 2018 as approved by the board of directors. Mr. Nathanielsz has also accrued unused annual and long service leave in the amounts of $46,187 (AUD) ($31,827 USD) and $135,795 (AUD) ($97,134 USD) for fiscal years 2020 and 2019, respectively, which are included in the total above. | |
| (3) | On March 16, 2018, our board of directors granted Mr. Nathanielsz a $300,000 AUD ($221,970 USD) cash bonus for accomplishments while servicing as our chief executive officer, of which $59,221 was paid in the year ended June 30, 2018 and the balance was paid in the year ended June 30, 2019. On May 14, 2019, our board of directors granted Mr. Nathanielsz a $460,000 AUD ($322,414 USD) cash bonus for accomplishments while servicing as our chief executive officer, of which $64,372 was paid in the year ended June 30, 2019 and a further $136,606 was paid in the year ended June 30, 2020. On July 13, 2020 the Board approved a bonus of $240,000 AUD ($165,384 USD) being equal to 60% of Mr Nathanielsz base salary. | |
| (4) | Under the Nathanielsz Employment Agreement, Mr. Nathanielsz receives a 9.5% contribution to a pension of which he is the beneficiary. In addition, pursuant to the Nathanielsz Employment Agreement, we may make a monthly payment to cover the costs relating to Mr. Nathanielsz use of a vehicle. For the fiscal years ended June 30, 2020 and 2019, $32,757 and $40,430, respectively, was paid to Mr. Nathanielsz for use of a vehicle. |
| 87 |
Narrative to Summary Compensation Table
Employment Agreement with James Nathanielsz
We and James Nathanielsz previously entered into an employment agreement on February 25, 2015 (the “Nathanielsz Employment Agreement”) setting forth the terms and conditions of Mr. Nathanielsz employment as our President and Chief Executive Officer. The Nathanielsz Employment Agreement also provided that Mr. Nathanielsz would serve as a member of our board of directors. The Nathanielsz Employment Agreement was scheduled to expire on February 25, 2018; however, the term of the Nathanielsz Employment Agreement automatically renews for successive one-year periods unless either party provides 30 days’ prior written notice of its intent not to renew. On May 14, 2019, we entered into the Amended and Restated Employment Agreement with Mr. Nathanielsz pursuant to which the Nathanielsz Employment Agreement was terminated. See below under “New Employment Agreement with James Nathanielsz” for information regarding such agreement.
The Nathanielsz Employment Agreement provided Mr. Nathanielsz with a base salary of $25,000 AUD per month ($300,000 AUD annually) and a monthly contribution to Mr. Nathanielsz’s pension equal to 9.5% of his monthly salary. Mr. Nathanielsz had the ability to convert any accrued but unpaid salary into common stock at the end of each fiscal year at a conversion price to be determined by Mr. Nathanielsz and our Company, which will in no event be lower than par value or higher than the closing bid price on the date of conversion. We also agreed to pay Mr. Nathanielsz an annual discretionary bonus in an amount up to 200% of his annual base salary, which bonus shall be determined by the Board and based upon the performance of our Company.
Mr. Nathanielsz is entitled to 20 days of annual leave and 10 days of paid sick leave. Mr. Nathanielsz is also entitled to participate in employee benefits plans, fringe benefits and perquisites maintained by the Company to the extent the Company provides similar benefits or perquisites (or both) to similarly situated executives of the Company.
In the event that the Company provides notice of non-renewal of the Nathanielsz Employment Agreement, the Company terminates Mr. Nathanielsz without cause (as defined in the Nathanielsz Employment Agreement) or Mr. Nathanielsz terminates his employment for good reason (as defined in the Nathanielsz Employment Agreement), the Company has agreed to pay Mr. Nathanielsz a severance payment in an amount equal to Mr. Nathanielsz’s base salary for the year of termination in addition to accrued but unpaid salary, reimbursement of expenses and certain other employee benefits as determined under the terms of the applicable plans (“Accrued Amounts”). In the event that Mr. Nathanielsz provides notice of non-renewal of the Nathanielsz Employment Agreement, the Company terminates Mr. Nathanielsz for cause or Mr. Nathanielsz terminates his employment without good reason, Mr. Nathanielsz is only entitled to the Accrued Amounts.
We agreed to indemnify Mr. Nathanielsz for any liabilities, costs and expenses incurred in the event that he is made a party to a proceeding due to his roles with our Company, other than any proceeding initiated by Mr. Nathanielsz or us relating to any dispute with respect to the Nathanielsz Employment Agreement or Mr. Nathanielsz’s employment. Under the terms of the Nathanielsz Employment Agreement, Mr. Nathanielsz was also subject to certain restrictive covenants, including a one-year non-compete.
| 88 |
On April 14, 2016, our board of directors approved Amendment No.1 to the Nathanielsz Employment Agreement to include a provision pursuant to which we agreed to pay Mr. Nathanielsz a monthly amount to cover the costs relating to Mr. Nathanielsz use of a vehicle. Also on April 14, 2016, our board of directors approved the payment of an annual bonus to Mr. Nathanielsz based on certain performance achievements in 2015 fiscal year in accordance with the terms of the Nathanielsz Employment Agreement. The bonus amount approved was $200,000 AUD (or 66.66% of Mr. Nathanielsz then current base salary).
On April 14, 2016 (the “Grant Date”), our board of directors granted 572 stock options with an exercise price of $3750 per share (market value of our common stock on the Grant Date), to Mr. Nathanielsz. 191 of such stock options vested on April 14, 2016, 191 of such stock options vest on April 14, 2017 (the first anniversary of the Grant Date) and 191 of such stock options shall vest on April 14, 2018 (the second anniversary of the Grant Date). These stock options expire on April 14, 2021. The fair value of the 286,000 options at the Grant Date is $1,962,440.
On August 15, 2016, our board of directors granted Mr. Nathanielsz a cash bonus in the amount of $250,000 USD (representing 83.33% of his annual base salary), of which $130,000 was paid in the fiscal year ended June 30, 2017. An additional $50,000 of this bonus was paid in the fiscal year ended June 30, 2018, pursuant to the terms of the Nathanielsz Employment Agreement, based upon the performance of our Company.
On September 25, 2017, we and Mr. Nathanielsz entered into an amendment to the Nathanielsz Employment Agreement. The amendment provided that the annual leave section of the Nathanielsz Employment Agreement be changed to permit any unused annual leave to roll over from year-to-year and that Mr. Nathanielsz would be entitled to receive any accrued but unpaid annual leave in the event of the termination of his employment. The amendment also acknowledged that Mr. Nathanielsz had accrued $121,884 of unused annual leave since he joined our Company in 2007. These amended provisions were intended to make the Nathanielsz Employment Agreement consistent with Australian law governing employee leave. In addition, the amendment clarified certain activities that Mr. Nathanielsz is prohibited from engaging in while employed with us in order to prevent competitive harm.
On March 16, 2018, our board of directors approved an increase of AU$100,000 (US$77,328.33) in Mr. Nathanielsz’ annual base salary, from AU$300,000 (US$231,984.99) to AU$400,000 (US$309,313.32), effective as of such date. In addition, having reviewed our corporate objectives and performance criteria, including short-term and long-term performance goals for Mr. Nathanielsz, our board of directors awarded a cash bonus of AU$300,000 (US$231,984.99) to Mr. Nathanielsz, which was equal to 100% of his annual base salary in the 2017 fiscal year, and was consistent with the bonus parameters set forth in the Nathanielsz Employment Agreement.
On May 14, 2019, the Board awarded a cash bonus of AU$460,000 (USD $322,414) to Mr. Nathanielsz, which is equal to 115% of his annual base salary in 2019 and is consistent with the bonus parameters set forth in Mr. Nathanielsz’s existing employment agreement with Company. On July 13, 2020 the Board approved a bonus of $240,000 AUD (USD $165,384) being equal to 60% of Mr Nathanielsz base salary.
| 89 |
Outstanding Equity Awards at Fiscal Year-End
The following table sets forth certain information with respect to grants of plan-based awards for the fiscal year ended June 30, 2020 to the Named Executive Officer. Except as set forth below, all of the outstanding equity awards granted to our Named Executive Officer were fully vested as of June 30, 2020.
| Option awards | Stock awards | |||||||||||||||||||||
| Name | Number
of Securities Underlying Unexercised Options (#) Exercisable |
Number
of Securities Underlying Unexercised Options (#) Unexercisable |
Option Exercise Price ($) |
Option Expiration Date |
Number
of Shares, Units or Other Rights That Have Not Vested (#) |
Market Value or Payout Value of Unearned Shares, Units or Other Rights That Have Not Vested ($) |
||||||||||||||||
| James Nathanielsz (1) | 572 | - | $ | 3,750 | April 14, 2021 | - | - | |||||||||||||||
| James Nathanielsz (2) | 13,000 | 26,000 | $ | 4.68 | May 13, 2029 | 39,000 | 165,757 | |||||||||||||||
| (1) | On April 14, 2016, the Board granted Mr. Nathanielsz 572 stock options at an exercise price of $3,750 per share (market value of the common stock on the Grant Date). 191 of such stock options vested on April 14, 2016 and expire on April 14, 2021, 191 of such stock options vested on April 14, 2017 (the first anniversary of the Grant Date) and expire on April 14, 2021 and 191 of such stock options vested on April 14, 2018 (the second anniversary of the Grant Date) and expire on April 14, 2021. The fair value of the 572 options at the Grant Date is $1,962,440. | |
| (2) | On May 14, 2019, the Board granted Mr. Nathanielsz 39,000 tenure based stock options at an exercise price of $4.68 per share and 78,000 performance based restricted stock units. The fair value of the 39,000 options and 78,000 restricted stock units at the grant date was $165,747 and $331,493, respectively. With 39,000 of such restricted stock vested on May 14, 2020 and the balance subject to performance conditions. |
Director Compensation for the Fiscal Year Ended June 30, 2020
| Name | Fees
earned or paid in cash ($) | Option
Awards ($) | All
Other Compensation ($) | Total
($) | ||||||||||||
| Dr. Julian Kenyon (1) | $ | 27,305 | (2) | $ | - | - | $ | 27,305 | ||||||||
| (1) | For purposes of the information included in the table, the conversion rate as of June 30, 2020, $0.6891 was used to convert amounts from AUD to USD. | |
| (2) | Under the Director Agreement (defined below), Dr. Kenyon received a gross consideration of $10,000 AUD per month through September 2016. Effective October 2016 Dr. Kenyon receives gross monthly compensation of $4,500 AUD or $3,218 USD per month for his services as a director of our Company. See “Compensation of Directors — Director Agreement with Julian Kenyon” below for additional details. |
Director Agreement with Dr. Julian Kenyon
The Director Agreement set forth the terms and conditions of Dr. Kenyon’s service as a director of our board of directors (the “Director Agreement”). Dr. Kenyon’s appointment term was originally for three years and was scheduled to expire on February 25, 2018; however, this term was scheduled to automatically renew for successive one-year periods unless either party provides 30 days’ prior written notice of its intent not to renew. On May 14, 2019, we entered into the Amended and Restated Services Agreement with Dr. Kenyon pursuant to which the Director Agreement was terminated. See below under “New Services Agreement with Julian Kenyon” for information regarding such agreement.
Under the Director Agreement (defined below), effective October 2016, Dr. Kenyon received gross monthly compensation of $4,500 AUD ($54,000 AUD annualized) or $3,489 USD per month for his services as a director of our Company. Dr. Kenyon had the ability to convert any accrued but unpaid compensation into common stock at the end of each fiscal year at a conversion price to be determined by Dr. Kenyon and us, which could not be lower than par value or higher than the closing bid price on the date of conversion.
The Company agreed to indemnify Dr. Kenyon for any liabilities, costs and expenses incurred in the event that he was made a party to a proceeding due to his role with our Company, other than any proceeding initiated by Dr. Kenyon or us relating to any dispute with respect to the Director Agreement or Dr. Kenyon’s service as a director. Under the terms of the Director Agreement, Dr. Kenyon was also subject to certain restrictive covenants, including a one-year non-compete.
| 90 |
On April 14, 2016 (the “Grant Date”), the board of directors of the Company granted 572 stock options with an exercise price of $3,750 per share (market value of the Company’s common stock on the Grant Date), to Dr. Kenyon. 191 of such stock options vested on April 14, 2016 and expire on April 14, 2021, 191 of such stock options vested on April 14, 2017 (the first anniversary of the Grant Date) and expire on April 14, 2021 and 191 of such stock options vested on April 14, 2018 (the second anniversary of the Grant Date) and expire on April 14, 2021. The fair value of the 572 options at the Grant Date was $1,962,440.
Effective October 2016, Dr. Kenyon received gross monthly compensation of $4,500 and on $3,489 USD per month.
Other Director Compensation
Our directors are reimbursed for reasonable expenses incurred in attending meetings and carrying out duties as board members.
Scientific Advisory Board Members Compensation
We have entered into Scientific Advisory Board Member Agreements with certain members of our Scientific Advisory Board (the “SAB Agreements”). The SAB Agreements contain substantially similar terms and primarily relate to the protection of our intellectual property. The SAB Agreements also include provisions for the members’ compensation for the services performed as a member of the Scientific Advisory Board. Messrs. Kutz, Brandt and Smyth each are paid a monetary fee for each year of service provided.
New Employment Agreement with James Nathanielsz
On May 14, 2019, we and James Nathanielsz entered into an Amended and Restated Employment Agreement (the “New Employment Agreement”) setting forth the new terms and conditions of Mr. Nathanielsz employment as our Chief Executive Officer and for his services as a director of our Company. The term of the New Employment Agreement is three years (the “Term”), subject to automatic 1-year renewals, at an annual salary of $400,000 AUD. Mr. Nathanielsz is also eligible to earn an annual fiscal year cash performance bonus for each fiscal year of his employment period with the Company in accordance with the Company’s annual bonus plan applicable to the Company’s senior executives. Mr. Nathanielsz’s “target” performance bonus shall be 200% of his average annualized base salary during the fiscal year for which the performance bonus is earned. Mr. Nathanielsz was also granted options (the “Nathanielsz Options”) to purchase 19,500,000 shares of our common stock, with an exercise price per share equal to 110% of the closing market price of our common stock on the date of approval of such grant by the Company’s Board of Directors (the “Board”), (ii) 19,500,000 restricted stock units of the Company (the “Initial Nathanielsz RSUs”), and (iii) an additional 19,500,000 restricted stock units of the Company (the “Additional Nathanielsz RSUs” and together with the Initial RSUs, the “Nathanielsz RSUs”). The Nathanielsz Options and the Nathanielsz RSUs were granted pursuant to the Company’s 2019 Equity Incentive Plan (the “Plan”) adopted by the Board on the Effective Date. The Nathanielsz Options have a term of 10 years from the date of grant. 1/3rd of the Nathanielsz Options shall vest every successive one-year anniversary following the Effective Date, provided, that on each such vesting date Mr. Nathanielsz is employed by the Company and subject to the other provisions of his New Employment Agreement. The Initial Nathanielsz RSUs shall vest on the one-year anniversary of the Effective Date, subject to Mr. Nathanielsz’s continued employment with the Company through such vesting date. The Additional Nathanielsz RSUs will vest as follows, subject to Mr. Nathanielsz’s continued employment with the Company through the applicable vesting date: (i) 3,900,000 of the Additional Nathanielsz RSUs shall vest upon the Company submitting Clinical Trial Application (the “CTA”) for PRP, the Company’s lead product candidate (“PRP”), for a First-In-Human study for PRP (the “Study”) in an applicable jurisdiction to be selected by the Company, (ii) 3,900,000 of the Additional Nathanielsz RSUs shall vest upon the CTA being approved in an applicable jurisdiction, (iii) 3,900,000 of the Additional RSUs shall vest upon the Company completing an equity financing in the amount of at least $4,000,000 in gross proceeds, including this offering, (iv) 3,900,000 of the Additional Nathanielsz RSUs shall vest upon the shares of our common stock being listed on a senior stock exchange (NYSE, NYSEMKT or NASDAQ), and (v) the remaining 3,900,000 of the Additional Nathanielsz RSUs shall vest upon the Company enrolling its first patient in the Study. Each vested Nathanielsz RSU shall be settled by delivery to Mr. Nathanielsz’s of one share of our common stock on the first to occur of: (i) the date of a Change of Control (as defined in the New Employment Agreement), (ii) the date that is ten business days following the vesting of such Nathanielsz RSU, (iii) the date of Mr. Nathanielsz’s death or Disability (as defined in his New Employment Agreement), and (iv) Mr. Nathanielsz’s employment being terminated either by the Company without Cause or by Mr. Nathanielsz for Good Reason (as defined in his New Employment Agreement). In the event of a “Change of Control” (as defined in his New Employment Agreement), any unvested portion of the Nathanielsz Options and the Nathanielsz RSUs shall vest immediately prior to such event. Each Nathanielsz RSU grant will be evidenced by an award agreement that shall specify such other terms and conditions as the Board, in its sole discretion, will determine in accordance with the terms and conditions of the 2019 Plan, including all terms, conditions and restrictions related to the grant and the form of payout, which, subject to the 2019 Plan, may be left to the discretion of the Board.
| 91 |
If Mr. Nathanielsz’s employment is terminated by the Company without “Cause” or by Mr. Nathanielsz for “Good Reason” (each as defined in his New Employment Agreement, subject to the Company’s right to cure), he will be entitled to termination benefits, pursuant to which (x) the Company will be obligated to pay Mr. Nathanielsz certain accrued obligations, any unpaid Prior Year Bonus and any Pro Rata Bonus (each as defined in his New Employment Agreement) for a period from the termination date through the lesser of 12 months or the period through and inclusive of the last day of the Term, (y) all of the unvested Nathanielsz Options and other equity awards (other than Nathanielsz RSUs) shall automatically accelerate and become vested and exercisable for a period of 6 months from the termination date or the date the award first becomes vested and exercisable, but in all events no later than the applicable term for each such award; and (z) all of the unvested Nathanielsz RSUs shall automatically and immediately become vested, as of the termination date, and such vested Nathanielsz RSUs shall be settled as set forth in the New Employment Agreement, and all restrictions on such equity awards shall automatically and immediately lapse. The New Employment Agreement contains customary restrictive covenants for the benefit of the Company, including a one-year non-compete provision, non-interference with the Company’s business after termination of employment and protection of the Company’s confidential information, certain customary representations and warranties and standard Company indemnification obligations.
New Services Agreement with Julian Kenyon
On May 14, 2019, we and Julian Kenyon entered into an Amended and Restated Services Agreement (the “Services Agreement”) setting forth the new terms and conditions of Dr. Kenyon’s employment as our Chief Scientific Officer and for his services as a director of our Company. The term of the Services Agreement is for three years, subject to automatic 1-year renewals, at an annual salary of $54,000 AUD. In connection with the execution of the Services Agreement, Dr. Kenyon was designated as an executive officer of the Company and assumed a more active role with the Company. Dr. Kenyon was also granted options (the “Kenyon Options”) to purchase 9,750,000 shares of our common stock, with an exercise price per share equal to 100% of the closing market price of the Common Stock on the date of approval of such grant by the Board, (ii) 9,750,000 restricted stock units of the Company (the “Initial Kenyon RSUs”), and (iii) an additional 9,750,000 restricted stock units of the Company (the “Additional Kenyon RSUs” and together with the Initial RSUs, the “Kenyon RSUs”). The Kenyon Options and the Kenyon RSUs were granted pursuant to the Plan. The Kenyon Options have a term of 10 years from the date of grant. 1/3rd of the Kenyon Options shall vest every successive one-year anniversary following the Effective Date, provided, that on each such vesting date Dr. Kenyon is employed by the Company and subject to the other provisions of his Services Agreement. The Initial Kenyon RSUs shall vest on the one-year anniversary of the Effective Date, subject to Dr. Kenyon’s continued employment with the Company through such vesting date. The Additional Kenyon RSUs will vest as follows, subject to Dr. Kenyon’s continued employment with the Company through the applicable vesting date: (i) 2,437,500 of the Additional Kenyon RSUs shall vest upon the Company submitting the CTA for PRP for the Study in an applicable jurisdiction to be selected by the Company, (ii) 2,437,500 of the Additional Kenyon RSUs shall vest upon the Company completing an equity financing in the amount of at least $4,000,000 in gross proceeds, including this offering, (iii) 2,437,500 of the Additional Kenyon RSUs shall vest upon the shares of the Company’s Common Stock being listed on a senior stock exchange (NYSE, NYSEMKT or NASDAQ), and (iv) the remaining 2,437,500 of the Additional Kenyon RSUs shall vest upon the Company enrolling its first patient in the Study. Each vested Kenyon RSU shall be settled by delivery to Dr. Kenyon’s of one share of Common Stock on the first to occur of: (i) the date of a Change of Control (as defined in the Services Agreement), (ii) the date that is ten business days following the vesting of such Kenyon RSU, (iii) the date of Dr. Kenyon’s death or Disability (as defined in the Services Agreement), and (iv) Dr. Kenyon’s employment being terminated either by the Company without Cause or by Dr. Kenyon for Good Reason (as defined in his Services Agreement). In the event of a “Change of Control” (as defined in his Services Agreement), 50% of any unvested portion of the Kenyon Options and the Kenyon RSUs shall vest immediately prior to such event. Each Kenyon RSU grant will be evidenced by an award agreement that shall specify such other terms and conditions as the Board, in its sole discretion, will determine in accordance with the terms and conditions of the 2019 Plan, including all terms, conditions and restrictions related to the grant and the form of payout, which, subject to the 2019 Plan, may be left to the discretion of the Board. Dr. Kenyon has the ability to convert any accrued but unpaid compensation into shares of our common stock at the end of each fiscal year at a conversion price to be determined by Dr. Kenyon and us, which will in no event be lower than par value or higher than the closing bid price on the date of conversion.
| 92 |
If Dr. Kenyon’s employment is terminated by the Company without “Cause” or by Dr. Kenyon for “Good Reason” (each as defined in his Services Agreement, subject to the Company’s right to cure), he will be entitled to termination benefits, pursuant to which (x) the Company will be obligated to pay Dr. Kenyon certain accrued obligations and severance in the form of salary payments for a period of 12 months, (y) 50% of the unvested Kenyon Options and other equity awards (other than Kenyon RSUs) shall automatically accelerate and become vested and exercisable for a period of 3 months from the termination date or the date the award first becomes vested and exercisable, but in all events no later than the applicable term for each such award; and (z) 50% of the unvested Kenyon RSUs shall automatically and immediately become vested, as of the termination date, and such vested Kenyon RSUs shall be settled as set forth in the Services Agreement, and all restrictions on such equity awards shall automatically and immediately lapse. The Services Agreement contains customary restrictive covenants for the benefit of the Company, including a two-year non-compete provision, non-interference with the Company’s business after termination of employment and protection of the Company’s confidential information, certain customary representations and warranties and standard Company indemnification obligations.
Narrative Disclosure of Compensation Policies and Practices as They Relate to Our Risk Management
We believe that our compensation policies and practices for all employees and other individual service providers, including executive officers, do not create risks that are reasonably likely to have a material adverse effect on us.
TRANSACTIONS WITH RELATED PERSONS, PROMOTERS AND CERTAIN CONTROL PERSONS AND
DIRECTOR INDEPENDENCE
The following includes a summary of transactions since July 1, 2018 to which we have been a party, in which the amount involved in the transaction exceeded $120,000, and in which any of our directors, executive officers or, to our knowledge, beneficial owners of more than 5% of our capital stock or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest, other than equity and other compensation, termination, change in control and other arrangements, which are described above under “Executive Compensation.”
Effective May 5, 2016, we entered into an agreement for the lease of our principal executive offices with North Horizon Pty Ltd, of which Mr. Nathanielsz and his wife are the owners and directors. The lease has a five-year term and provides for annual rental payments of $39,600 AUD, which includes $3,600 of goods and service tax, for total payments of $198,000 AUD during the term of the lease.
Mr. Nathanielsz’s wife, Sylvia Nathanielsz, is and has been an employee of our Company since October 2015. Effective February 1, 2018, Mrs. Nathanielsz receives an annual salary of $120,000 AUD, or $80,904 USD, and is entitled to benefits customarily expected to be provided to employees of the Company. From July 2015 until October 2015, Mrs. Nathanielsz was an independent contractor serving our Company and was paid approximately $13,632 for her services.
| 93 |
Employment and Director Compensation Arrangements
The relationships and related party transactions described herein are in addition to any employment and director compensation arrangements with our executive officers and directors, which are described above under “Executive Compensation — Narrative to Summary Compensation Table and Director Compensation,” “Executive Compensation — New Employment Agreement with James Nathanielsz” and “Executive Compensation — New Services Agreement with Julian Kenyon.”
Indemnification Agreements
Our Certificate of Incorporation provides that none of our officers or directors shall be personally liable for any obligations of our Company or for any duties or obligations arising out of any acts or conduct of said officer or director performed for or on behalf of our Company, including without limitation, acts of negligence or contributory negligence. In addition, our Certificate of Incorporation provides that we shall indemnify and hold harmless each person and their heirs and administrators who shall serve at any time hereafter as a director or officer of our Company from and against any and all claims, judgments and liabilities to which such persons shall become subject by reason of their having heretofore or hereafter been a director or officer of our Company, or by reason of any action alleged to have heretofore or hereafter taken or omitted to have been taken by him or her as such director or officer, and that we shall reimburse each such person for all legal and other expenses reasonably incurred by him or her in connection with any such claim, judgment or liability, including our power to defend such persons from all suits or claims as provided for under the provisions of the General Corporation Law of the State of Delaware; provided, however, that no such persons shall be indemnified against, or be reimbursed for, any expense incurred in connection with any claim or liability arising out of his (or her) own willful misconduct. Our Certificate of Incorporation also provide that we are obligated to advance expenses incurred by a director or officer in advance of the final disposition of any action or proceeding, and permit us to secure insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in that capacity regardless of whether we would otherwise be permitted to indemnify him or her under the provisions of Delaware law. In addition, we intend to enter into indemnification agreements with our directors and officers and some of our executives may have certain indemnification rights arising under their employment agreements with us. We believe that these provisions of our Certificate of Incorporation and indemnification agreements are necessary to attract and retain qualified persons as directors and officers.
The limitation of liability and indemnification provisions in our Certificate of Incorporation may discourage stockholders from bringing a lawsuit against our directors for breach of their fiduciary duties. They may also reduce the likelihood of derivative litigation against our directors and officers, even though an action, if successful, might benefit us and our stockholders. A stockholder’s investment may be harmed to the extent we pay the costs of settlement and damage awards against directors and officers pursuant to these indemnification provisions.
On May 14, 2019, our board of directors approved a form of Indemnification Agreement (“Indemnification Agreement”) for each of our officers and directors. The Indemnification Agreement requires us to indemnify our directors and officers and to advance expenses on behalf of such directors or officers to the fullest extent permitted by applicable law and establish the procedures by which a director or executive officer may request and receive indemnification. The Indemnification Agreement is in addition to other rights to which a director or officer may be entitled under our Certificate of Incorporation, Bylaws and applicable law.
Policies and Procedures for Transactions with Related Persons
We intend to adopt a written related-person transactions policy that will set forth our policies and procedures regarding the identification, review, consideration and oversight of “related-person transactions.” For purposes of this policy only, a “related-person transaction” shall be a transaction, arrangement or relationship (or any series of similar transactions, arrangements or relationships) in which we and any “related person” are participants involving an amount that exceeds $120,000.
Director Independence
Our board of directors has reviewed the independence of our directors and has determined that none of our directors qualifies as an independent director pursuant to Rule 5605(a)(2) of Nasdaq and applicable SEC rules and regulations. In making this determination, our board of directors considered the relationships that each of our directors has with us and all other facts and circumstances our board of directors deemed relevant in determining their independence.
| 94 |
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following sets forth information as of September 23, 2020 regarding the number of shares of our Common Stock beneficially owned by (i) each person that we know beneficially owns more than 5% of our outstanding Common Stock, (ii) each of our directors and named executive officer and (iii) all of our directors and named executive officers as a group.
The amounts and percentages of our Common Stock beneficially owned are reported on the basis of SEC rules governing the determination of beneficial ownership of securities. Under the SEC rules, a person is deemed to be a “beneficial owner” of a security if that person has or shares “voting power,” which includes the power to vote or to direct the voting of such security, or “investment power,” which includes the power to dispose of or to direct the disposition of such security. A person is also deemed to be a beneficial owner of any securities of which that person has the right to acquire beneficial ownership within 60 days through the exercise of any stock option, warrant or other right, and the conversion of preferred stock. Under these rules, more than one person may be deemed a beneficial owner of the same securities and a person may be deemed to be a beneficial owner of securities as to which such person has no economic interest. Unless otherwise indicated, each of the shareholders named in the table below, or his or her family members, has sole voting and investment power with respect to such shares of our Common Stock. Except as otherwise indicated, the address of each of the shareholders listed below is: c/o Propanc Biopharma, Inc., 302, 6 Butler Street, Camberwell, VIC, 3124 Australia.
| Common
Stock Beneficially Owned |
Series
A Preferred Stock Beneficially Owned |
Series
B Preferred Stock Beneficially Owned |
||||||||||||||||||||||
| Name and Address of Beneficial Owner | Number of Shares Beneficially Owned | Percentage
of Class(1) |
Number of Shares Beneficially Owned | Percentage
of Class(2) |
Number of Shares Beneficially Owned | Percentage
of Class (2) |
||||||||||||||||||
| North Horizon Pty Ltd.(3) | 345 | * | 500,000 | 100 | % | - | - | |||||||||||||||||
| James Nathanielsz(4) | 917 | * | - | - | 1 | 100 | % | |||||||||||||||||
| Dr. Julian Kenyon(5) | 802 | * | - | - | - | - | ||||||||||||||||||
| All directors and executive officers, as a group (2 persons) | 1,719 | * | 500,000 | 100 | % | 1 | 100 | % | ||||||||||||||||
* Represents beneficial ownership of less than one percent.
(1) Applicable percentages are based on 668,670,618 shares of our common stock outstanding as of September 28, 2020.
(2) Applicable percentages are based on 500,000 shares of our Series A Preferred Stock and 1 share of our Series B Preferred Stock outstanding as of September 28, 2020, except where the person or entity has the right to receive shares within the next 60 days, which would increase the number of shares owned by such person or entity and the number of shares outstanding.
(3) North Horizon Pty Ltd. is a Nathanielsz Family Trust. Mr. James Nathanielsz, the Chief Executive Officer and a director of our Company, has voting and investment power over these shares.
(4) Represents 345 shares of our common stock held by North Horizon Pty Ltd. and 572 shares of common stock issuable to Mr. Nathanielsz pursuant to his stock options currently exercisable on or before April 14, 2021. Excludes 26,000 options, 39,000 restricted stock units that vested in May, 2020 and 39,000 restricted stock units that are subject to certain vesting conditions, as discussed above in the section captioned “Executive Compensation — New Employment Agreement with James Nathanielsz,”.
(5) Represents 230 shares of our common stock held by Dr. Julian Kenyon, a director of our Company, and 572 shares of our common stock issuable to Dr. Kenyon pursuant to his stock options currently exercisable on or before April 14, 2021. Excludes 6,500 options and 19,500 restricted stock units that vested in in May 2020, and 19,500 restricted stock units that are subject to certain vesting conditions, as discussed above in the section captioned “Executive Compensation — New Services Agreement with Julian Kenyon”.
The following is a brief description of the securities we are offering. This summary does not purport to be complete in all respects. This description is subject to and qualified entirely by the terms of our Certificate of Incorporation, as amended (the “Certificate of Incorporation”), our Bylaws, and the General Corporation Law of the State of Delaware. Copies of our Certificate of Incorporation and Bylaws have been filed as exhibits to the registration statement of which this prospectus is a part. Also see “Where You Can Find More Information” section of this prospectus.
We are offering (i) 150,000,000 shares, which consists of 804,518 shares of common stock and 149,195,482 warrants to purchase shares. The shares of common stock and warrants included in this offering will be issued separately. Neither the shares nor the warrants will be issued or certificated. We are also registering the shares of common stock included in this offering and the shares of common stock issuable from time to time upon exercise of the warrants included in this offering.
Authorized Capital Stock
Our authorized capital stock consists of 1,000,000,000 shares of common stock, $0.001 par value per share, and 1,500,005 shares of preferred stock, $0.01 par value per share. As of September 28, 2020, there were (i) 668,670,618 shares of our common stock issued and outstanding and (ii) 500,001 shares of our preferred stock issued and outstanding, consisting of 500,000 preferred shares designated as our Series A Preferred Stock and one preferred share designated as our Series B Preferred Stock.
As of September 28, 2020, we had 74 holders of record of our common stock, which excludes stockholders whose shares were held in nominee or street name by brokers. The actual number of common stockholders is greater than the number of record holders and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers and other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
Common Stock
Voting ― Holders of our common stock are entitled to one vote for each share held of record on all matters submitted to a vote of the stockholders, including the election of directors, and do not have cumulative voting rights. Accordingly, the holders of a majority of the shares of our common stock entitled to vote in any election of directors can elect all of the directors standing for election.
| 95 |
Dividends ― Subject to preferences that may be applicable to any then outstanding preferred stock, the holders of common stock are entitled to receive dividends, if any, as may be declared from time to time by our board of directors out of legally available funds.
Liquidation ― In the event of our liquidation, dissolution or winding up, holders of our common stock will be entitled to share ratably in the net assets legally available for distribution to stockholders after the payment of all of our debts and other liabilities, subject to the satisfaction of any liquidation preference granted to the holders of any outstanding shares of preferred stock.
Rights and Preferences ― Holders of our common stock have no preemptive, conversion or subscription rights, and there are no redemption or sinking fund provisions applicable to our common stock. The rights, preferences and privileges of the holders of our common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of our preferred stock that we may designate and issue in the future.
Fully Paid and Nonassessable ― All of our outstanding shares of common stock are, and the shares of common stock to be issued in this offering will be, fully paid and nonassessable.
Preferred Stock
Our board of directors has the authority, without further action by the stockholders, to issue up to 1,500,005 shares of preferred stock in one or more series, to establish from time to time the number of shares to be included in each such series, to fix the rights, preferences and privileges of the shares of each wholly unissued series and any qualifications, limitations or restrictions thereon and to increase or decrease the number of shares of any such series, but not below the number of shares of such series then outstanding.
Our board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of the common stock. The issuance of preferred stock, while providing flexibility in connection with possible acquisitions and other corporate purposes, could, among other things, have the effect of delaying, deferring or preventing a change in our control that may otherwise benefit holders of our common stock and may adversely affect the market price of the common stock and the voting and other rights of the holders of common stock.
As of June 30, 2020, 500,001 shares of preferred stock were issued and outstanding, consisting of: (i) 500,000 preferred shares designated as Series A Preferred Stock (the “Series A Preferred Stock”) pursuant to our Certificate of Designation filed with the Secretary of State of the State of Delaware on December 9, 2014; and (ii) one preferred share designated as Series B Preferred Stock (the “Series B Preferred Stock”) pursuant to our Certificate of Designation filed with the Secretary of State of the State of Delaware on June 16, 2015. Up to five shares have been designated as Series B Preferred Stock.
Mr. Nathanielsz, our Chief Executive Officer, Chairman, Secretary, Treasurer and a director, beneficially owns all of the shares of Series A Preferred Stock via North Horizon Pty Ltd., which entitles him, as a holder of Series A Preferred Stock, to vote on all matters submitted or required to be submitted to a vote of our stockholders, except election and removal of directors, and each share of Series A Preferred Stock entitles him to 0.004 votes per each outstanding share of Series A Preferred Stock. North Horizon Pty Ltd. is a Nathanielsz Family Trust. Mr. Nathanielsz has voting and investment power over these shares.
Mr. Nathanielsz directly beneficially owns such one share of Series B Preferred Stock. Each holder of outstanding shares of Series B Preferred Stock is entitled to voting power equivalent to the number of votes equal to the total number of shares of common stock outstanding as of the record date for the determination of stockholders entitled to vote at each meeting of stockholders of our Company and entitled to vote on all matters submitted or required to be submitted to a vote of the stockholders of the Company.
All of our issued and outstanding shares of preferred stock have no rights to dividends, profit sharing or liquidation preferences.
| 96 |
Authorized and Unissued Capital Stock
Delaware law does not require stockholder approval for any issuance of authorized shares. These additional shares may be used for a variety of corporate purposes, including future public offerings, to raise additional capital or to facilitate acquisitions.
One of the effects of the existence of unissued and unreserved common stock or preferred stock may be to enable our board of directors to issue shares to persons friendly to current management, which issuance could render more difficult or discourage an attempt to obtain control of our company by means of a merger, tender offer, proxy contest or otherwise, and thereby protect the continuity of our management and possibly deprive the stockholders of opportunities to sell their shares at prices higher than prevailing market prices.
Warrants
Outstanding Warrants
As of August 7, 2020, there were 151,170,514 warrants outstanding and exercisable with expiration dates commencing August 2020 and continuing through August 2024, with a weighted average exercise price per share of $0.15
Warrants Included in Offering
The following summary of certain terms and provisions of warrants included that are being offered hereby is not complete and is subject to, and qualified in its entirety by, the provisions of the warrants, the form of which is filed as an exhibit to the registration statement of which this prospectus forms a part. Prospective investors should carefully review the terms and provisions of the form of warrant for a complete description of the terms and conditions of the warrants.
Pre-Funded Warrants
The following summary of certain terms and provisions of pre-funded warrants included in the pre-funded units that are being offered hereby is not complete and is subject to, and qualified in its entirety by, the provisions of the pre-funded warrant, the form of which is filed as an exhibit to the registration statement of which this prospectus forms a part. Prospective investors should carefully review the terms and provisions of the form of pre-funded warrant for a complete description of the terms and conditions of the pre-funded warrants.
Duration and Exercise Price
Each pre-funded warrant will have an initial exercise price per share equal to $0.0001. The pre-funded warrants will be immediately exercisable and may be exercised at any time until the pre-funded warrants are exercised in full. The exercise price and number of shares of common stock issuable upon exercise is subject to appropriate adjustment in the event of stock dividends, stock splits, reorganizations or similar events affecting our common stock and the exercise price. The pre-funded warrants will be issued separately from the accompanying common warrants included in the pre-funded units, and may be transferred separately immediately thereafter.
Exercisability
The pre-funded warrants will be exercisable, at the option of each holder, in whole or in part, by delivering to us a duly executed exercise notice accompanied by payment in full for the number of shares of our common stock purchased upon such exercise (except in the case of a cashless exercise as discussed below). A holder (together with its affiliates) may not exercise any portion of the pre-funded warrant to the extent that the holder would own more than 4.99% of the outstanding common stock immediately after exercise, except that upon at least 61 days’ prior notice from the holder to us, the holder may increase the amount of ownership of outstanding stock after exercising the holder’s pre-funded warrants up to 9.99% of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the pre-funded warrants. Purchasers of pre-funded units in this offering may also elect, prior to the issuance of the pre funded warrants, to have the initial exercise limitation set at 9.99% of our outstanding common stock.
| 97 |
Cashless Exercise
If, at the time a holder exercises its pre-funded warrants, a registration statement registering the issuance of the shares of common stock underlying the pre-funded warrants under the Securities Act is not then effective or available for the issuance of such shares, then in lieu of making the cash payment otherwise contemplated to be made to us upon such exercise in payment of the aggregate exercise price, the holder may elect instead to receive upon such exercise (either in whole or in part) the net number of shares of common stock determined according to a formula set forth in the pre-funded warrants.
Transferability
Subject to applicable laws, a pre-funded warrant may be transferred at the option of the holder upon surrender of the pre-funded warrant to us together with the appropriate instruments of transfer.
Fractional Shares
No fractional shares of common stock will be issued upon the exercise of the pre-funded warrants. Rather, the number of shares of common stock to be issued will, at our election, either be rounded up to the nearest whole number or we will pay a cash adjustment in respect of such final fraction in an amount equal to such fraction multiplied by the exercise price.
Trading Market
There is no trading market available for the pre-funded warrants on any securities exchange or nationally recognized trading system.
Right as a Stockholder
Except as otherwise provided in the pre-funded warrants or by virtue of such holder’s ownership of shares of our common stock, the holders of the pre-funded warrants do not have the rights or privileges of holders of our common stock, including any voting rights, until they exercise their pre-funded warrants.
Common Stock Warrants
Duration and Exercise Price
The Series A Warrants have an exercise price equal to $0.20 and will expire on the three (3) year anniversary of the date they first become exercisable. The Series B Warrants have an exercise price equal to $0.04 and will expire on the three (3) year anniversary of the date they first become exercisable. The Series C Warrant has an exercise price equal to $0.20 and will expire on the three (3) year anniversary of the date they first become exercisable.
Cashless Exercise
If, at the time a holder exercises the Series A Warrants, Series B Warrants or Series C Warrants (the “Warrants”), a registration statement registering the issuance of the shares of common stock underlying the Warrants under the Securities Act is not then effective or available for the issuance of such shares, then in lieu of making the cash payment otherwise contemplated to be made to us upon such exercise in payment of the aggregate exercise price, the holder may elect instead to receive upon such exercise (either in whole or in part) the net number of shares of common stock determined according to a formula set forth in the Warrants.
Exercisability
The Warrants will be exercisable, at the option of each holder, in whole or in part, by delivering to us a duly executed exercise notice accompanied by payment in full for the number of our shares of common stock purchased upon such exercise (except in the case of a cashless exercise as discussed below). A holder (together with its affiliates) may not exercise any portion of the Warrant to the extent that the holder would own more than 4.99% (or, at the election of a purchaser prior to issuance of the warrant, 9.99%) of the outstanding shares of common stock immediately after exercise, except that upon at least 61 days’ prior notice from the holder to us, the holder may increase the amount of ownership of outstanding shares after exercising the holder’s warrants up to 9.99% of the number of our shares of common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the Warrants.
| 98 |
Fractional Shares
No fractional shares of common stock will be issued upon the exercise of the Warrants. Rather, the number of shares of common stock to be issued will, at our election, either be rounded up to the nearest whole number or we will pay a cash adjustment in respect of such final fraction in an amount equal to such fraction multiplied by such warrant’s exercise price.
Transferability
Subject to applicable laws, the Warrant may be transferred at the option of the holder upon surrender of the warrant to us together with the appropriate instruments of transfer.
Exchange Listing
We do not intend to list the Warrants on any securities exchange or nationally recognized trading system.
Rights as a Shareholder
Except as otherwise provided in the Warrants or by virtue of such holder’s ownership of our shares of common stock, the holders of the Warrants do not have the rights or privileges of holders of our shares of common stock, including any voting rights, until they exercise their warrants.
Fundamental Transaction
In the event of a fundamental transaction which is approved by our Board, the holders of the Warrants have the right to require us or a successor entity to redeem the Warrant for cash in the amount of the Black-Scholes value of the unexercised portion of the warrant on the date of the consummation of the fundamental transaction. In the event of a fundamental transaction which is not approved by our Board, the holders of the Warrants have the right to require us or a successor entity to redeem the Warrant for the consideration paid in the fundamental transaction in the amount of the Black Scholes value of the unexercised portion of the warrant on the date of the consummation of the fundamental transaction.
| 99 |
Restricted Stock Units
As of June 30, 2020, pursuant to employment agreements dated in May 2019, the Company granted an aggregate of 78,000 and 39,000 restricted stock unit to the Company’s Chief Executive Officer and Chief Scientific Officer, respectively. The total 117,000 restricted stock units are subject to vesting terms as defined in the employment agreements and summarized above in the “Executive Compensation” section. The 117,000 restricted stock units were valued at the fair value of $4.25 per unit or $497,240 based on the quoted trading price on the date of grant.
Each vested restricted stock unit shall be settled by delivery to the holder thereof of one share of our common stock and/or the cash fair market value of such share of common stock on the date of settlement.
Options
As of June 30, 2020, the Company had entered into agreements to grant options to purchase 59,644 shares of its common stock, with a weighted average exercise price per share of $76.37.
Pursuant to employment agreements dated in May 2019, the Company granted options to purchase 39,000 and 19,500 shares of the Company’s common stock to the Company’s Chief Executive Officer and Chief Scientific Officer, respectively. The total 58,500 options have a term of 10 years from the date of grant and exercise price ranging from $4.25 to $4.675 per share. 1/3rd of these options shall vest every successive one-year anniversary, provided, that on each such vesting date, the Chief Executive Officer and Chief Scientific Officer are employed by the Company and subject to the other provisions of the employment agreement and summarized above in the “Executive Compensation” section. The 58,500 stock options were valued using a Black-Scholes model with the following assumptions: stock price at valuation date of $4.25 based on quoted trading price on date of grant, exercise price of $4.65, dividend yield of zero, years to maturity of 10.00, a risk free rate of 2.42%, and expected volatility 268% for a total value of $248,620.
Dividends
We have not paid any cash dividends to our stockholders. The declaration of any future cash dividends is at the discretion of our board of directors and depends upon our earnings, if any, our capital requirements and financial position, and general economic conditions. It is our present intention not to pay any cash dividends in the foreseeable future, but rather to reinvest earnings, if any, in our business operations.
Delaware Anti-Takeover Statute
We are subject to the provisions of Section 203 of the General Corporation Law of the State of Delaware regulating corporate takeovers. In general, Section 203 prohibits a publicly held Delaware corporation from engaging, under certain circumstances, in a business combination with an interested stockholder for a period of three years following the date the person became an interested stockholder unless:
| ● | prior to the date of the transaction, our board of directors approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder; | |
| ● | upon completion of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the voting stock outstanding, but not the outstanding voting stock owned by the interested stockholder, (1) shares owned by persons who are directors and also officers and (2) shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or | |
| ● | at or subsequent to the date of the transaction, the business combination is approved by our board of directors and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock which is not owned by the interested stockholder. |
| 100 |
Generally, a business combination includes a merger, asset or stock sale, or other transaction resulting in a financial benefit to the interested stockholder. An interested stockholder is a person who, together with affiliates and associates, owns or, within three years prior to the determination of interested stockholder status, did own 15% or more of a corporation’s outstanding voting stock. We expect the existence of this provision to have an anti-takeover effect with respect to transactions our board of directors does not approve in advance. We also anticipate that Section 203 may discourage attempts that might result in a premium over the market price for the shares of common stock held by stockholders.
The provisions of Delaware law and the provisions of our Certificate of Incorporation and Bylaws could have the effect of discouraging others from attempting hostile takeovers and, as a consequence, they might also inhibit temporary fluctuations in the market price of our common stock that often result from actual or rumored hostile takeover attempts. These provisions might also have the effect of preventing changes in our management. It is also possible that these provisions could make it more difficult to accomplish transactions that stockholders might otherwise deem to be in their best interests.
Bylaws
Provisions of our Bylaws may delay or discourage transactions involving an actual or potential change in our control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares or transactions that our stockholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our common stock. Among other things, our Bylaws:
| ● | permit our board of directors to issue up to 1,500,005 shares of our preferred stock, with any rights, preferences and privileges as they may designate (including the right to approve an acquisition or other change in our control); | |
| ● | provide that the authorized number of directors may be changed only by resolution of the board of directors; | |
| ● | provide that all vacancies, including newly created directorships, may, except as otherwise required by law, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum; and | |
| ● | do not provide for cumulative voting rights (therefore allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election, if they should so choose). |
The amendment of any of these provisions, with the exception of the ability of our board of directors to issue shares of preferred stock and designate any rights, preferences and privileges thereto, would require approval by the holders of a majority of our then outstanding common stock.
EXPERTS
Our consolidated financial statements as of and for the years ended June 30, 2020 and 2019, appearing in this prospectus and the registration statement of which it is a part, have been audited by Salberg & Company, P.A., an independent registered public accounting firm, as set forth in their report dated October 1, 2020 (which contains an explanatory paragraph regarding our ability to continue as a going concern) appearing elsewhere herein, and are included in reliance upon such report given on the authority of such firm as experts in accounting and auditing.
LEGAL MATTERS
The validity of the issuance of the Common Stock underlying the Units and Warrants hereby will be passed upon for us by Lucosky Brookman LLP.
| 101 |
QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable to smaller reporting companies.
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
There have been no changes in our independent registered public accounting firm during the last two fiscal years, and we have not had any material disagreements with our independent registered public accounting firm during that time.
WHERE YOU CAN FIND MORE INFORMATION
We have filed a registration statement on Form S-1 with the SEC covering the shares and warrants we are offering by this prospectus. This prospectus does not include all of the information contained in the registration statement. You should refer to the registration statement and its exhibits for additional information. Whenever we make reference in this prospectus to any of our contracts, agreements or other documents, the references are not necessarily complete and you should refer to the exhibits filed or documents incorporated by reference as part of the registration statement for copies of the actual contract, agreement or other document.
We file annual, quarterly and other periodic reports, proxy statements and other information with the SEC. You can read our SEC filings, including this registration statement, over the Internet at the SEC’s website at www.sec.gov. You may also read and copy any document we file with the SEC at its public reference facilities at 100 F Street NE, Washington, D.C. 20549. You may also obtain copies of these documents at prescribed rates by writing to the Public Reference Section of the SEC at 100 F Street NE, Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for further information on the operation of the public reference facilities.
Our Internet address is www.propanc.com. There we make available free of charge, on or through the investor relations section of our website, Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and amendments to those reports filed pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after we electronically file such material with the SEC. The information found on our website is not part of this prospectus and investors should not rely on any such information in deciding whether to invest.
Statements contained in this prospectus as to the contents of any contract or other document that we have filed as an exhibit to the registration statement are qualified in their entirety by reference to the exhibits for a complete statement of their terms and conditions.
The representations, warranties and covenants made by us in any agreement that is filed as an exhibit to the registration statement of which this prospectus is a part were made solely for the benefit of the parties to such agreement, including, in some cases, for the purpose of allocating risk among the parties to such agreements, and should not be deemed to be a representation, warranty or covenant to you. Moreover, such representations, warranties or covenants were made as of an earlier date. Accordingly, such representations, warranties and covenants should not be deemed as accurately representing the current state of our affairs.
| 102 |
DISCLOSURE OF COMMISSION POSITION ON INDEMNIFICATION FOR SECURITIES ACT LIABILITIES
Our Certificate of Incorporation contains provisions that limit the liability of our directors for monetary damages to the fullest extent permitted by Delaware law. Consequently, our directors will not be personally liable to us or our stockholders for monetary damages for any breach of fiduciary duties as directors, except liability for:
| ● | any breach of the director’s duty of loyalty to us or our stockholders; | |
| ● | any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; | |
| ● | unlawful payments of dividends or unlawful stock repurchases or redemptions as provided in Section 174 of the General Corporation Law of the State of Delaware; or | |
| ● | any transaction from which the director derived an improper personal benefit. |
Our Certificate of Incorporation, as amended, provides that we are required to indemnify our directors and officers, in each case to the fullest extent permitted by Delaware law. Our Certificate of Incorporation also provides that we are obligated to advance expenses incurred by a director or officer in advance of the final disposition of any action or proceeding, and permit us to secure insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in that capacity regardless of whether we would otherwise be permitted to indemnify him or her under the provisions of Delaware law.
To the extent that indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling our company pursuant to the foregoing provisions, we have been informed that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
| 103 |
| Page | |
| Audited Consolidated Financial Statements for the Fiscal Years Ended June 30, 2020 and 2019 | |
| Report of Independent Registered Public Accounting Firm | F-2 |
| Consolidated Balance Sheets as of June 30, 2020 and 2019 | F-3 |
| Consolidated Statements of Operations and Comprehensive Income (Loss) for the years ended June 30, 2020 and 2019 | F-4 |
| Consolidated Statements of Changes in Stockholders’ Deficit for the years ended June 30, 2020 and 2019 | F-5 |
| Consolidated Statements of Cash Flows for the years ended June 30, 2020 and 2019 | F-6 |
| Notes to Consolidated Financial Statements | F-7 |
| F-1 |

Report of Independent Registered Public Accounting Firm
To the Stockholders’ and the Board of Directors of:
Propanc Biopharma, Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of Propanc Biopharma, Inc. and Subsidiary (the “Company”) as of June 30, 2020 and 2019, the related consolidated statements of operations and comprehensive income (loss), changes in stockholders’ deficit, and cash flows, for each of the two years in the period ended June 30, 2020, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the consolidated financial position of the Company as of June 30, 2020 and 2019, and the consolidated results of its operations and its cash flows for each of the two years in the period ended June 30, 2020, in conformity with accounting principles generally accepted in the United States of America.
Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the consolidated financial statements, the Company has a net loss and cash used in operations of $4,740,723 and $1,849,589, respectively, in 2020 and has a working capital deficit, stockholders’ deficit and accumulated deficit of $3,670,921, $3,641,425 and $55,781,770, respectively, at June 30, 2020. These matters raise substantial doubt about the Company’s ability to continue as a going concern. Management’s Plan in regards to these matters is also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
| /s/ Salberg & Company, P.A. | |
| SALBERG & COMPANY, P.A. | |
| We have served as the Company’s auditor since 2011. | |
| Boca Raton, Florida | |
| October 1, 2020 |
2295 NW Corporate Blvd., Suite 240 • Boca Raton, FL 33431
Phone: (561) 995-8270 • Toll Free: (866) CPA-8500 • Fax: (561) 995-1920
www.salbergco.com • info@salbergco.com
Member National Association of Certified Valuation Analysts • Registered with the PCAOB
Member CPAConnect with Affiliated Offices Worldwide • Member AICPA Center for Audit Quality
| F-2 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
| June 30, 2020 | June 30, 2019 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS: | ||||||||
| Cash | $ | 67,007 | $ | 2,394 | ||||
| GST tax receivable | 2,015 | 5,439 | ||||||
| Prepaid expenses and other current assets | - | 83,299 | ||||||
| TOTAL CURRENT ASSETS | 69,022 | 91,132 | ||||||
| Security deposit - related party | 2,067 | 2,103 | ||||||
| Operating lease right-of-use assets, net - related party | 21,682 | - | ||||||
| Property and equipment, net | 5,747 | 8,417 | ||||||
| TOTAL ASSETS | $ | 98,518 | $ | 101,652 | ||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | ||||||||
| CURRENT LIABILITIES: | ||||||||
| Accounts payable | $ | 842,156 | $ | 917,337 | ||||
| Accrued expenses and other payables | 702,231 | 723,042 | ||||||
| Convertible notes and related accrued interest and premiums, net of discounts | 1,557,734 | 1,657,377 | ||||||
| Operating lease liability - related party | 25,072 | - | ||||||
| Embedded conversion option liabilities | 177,009 | 698,264 | ||||||
| Due to former director - related parties | 30,639 | 31,164 | ||||||
| Loans from directors and officer - related parties | 50,993 | 51,867 | ||||||
| Employee benefit liability | 354,109 | 323,837 | ||||||
| TOTAL CURRENT LIABILITIES | 3,739,943 | 4,402,888 | ||||||
| TOTAL LIABILITIES | $ | 3,739,943 | $ | 4,402,888 | ||||
| Commitments and Contingencies (See Note 9) | ||||||||
| STOCKHOLDERS’ DEFICIT: | ||||||||
| Preferred stock, 1,500,005 shares authorized, $0.01 par value: | ||||||||
| Series A preferred stock, $0.01 par value; 500,000 shares authorized; 500,000 shares issued and outstanding as of June 30, 2020 and 2019 | $ | 5,000 | $ | 5,000 | ||||
| Series B preferred stock, $0.01 par value; 5 shares authorized; 1 share issued and outstanding as of June 30, 2020 and 2019 | - | - | ||||||
| Common stock, $0.001 par value; 1,000,000,000 shares authorized; 258,120,381 and 968,042 shares issued; 258,120,332 and 967,993 outstanding as of June 30, 2020 and 2019, respectively | 258,120 | 968 | ||||||
| Additional paid-in capital | 50,656,031 | 45,713,322 | ||||||
| Accumulated other comprehensive income | 1,267,671 | 1,066,998 | ||||||
| Accumulated deficit | (55,781,770 | ) | (51,041,047 | ) | ||||
| Treasury stock (49 shares) | (46,477 | ) | (46,477 | ) | ||||
| TOTAL STOCKHOLDERS’ DEFICIT | (3,641,425 | ) | (4,301,236 | ) | ||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ DEFICIT | $ | 98,518 | $ | 101,652 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-3 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE INCOME (LOSS)
| Years Ended June 30, | ||||||||
| 2020 | 2019 | |||||||
| REVENUE | ||||||||
| Revenue | $ | - | $ | - | ||||
| OPERATING EXPENSES | ||||||||
| Administration expenses | 3,281,464 | 2,326,350 | ||||||
| Occupancy expenses | 32,809 | 28,062 | ||||||
| Research and development | 179,987 | 260,335 | ||||||
| TOTAL OPERATING EXPENSES | 3,494,260 | 2,614,747 | ||||||
| LOSS FROM OPERATIONS | (3,494,260 | ) | (2,614,747 | ) | ||||
| OTHER INCOME (EXPENSE) | ||||||||
| Interest expense | (1,748,381 | ) | (1,314,539 | ) | ||||
| Interest income | 946 | 31 | ||||||
| Other income | 57,636 | - | ||||||
| Derivative income (expense) | 385,293 | (2,472,146 | ) | |||||
| Gain on debt settlements, net | - | 14,101 | ||||||
| Gain on extinguishment of debt, net | 67,123 | 1,204,242 | ||||||
| Foreign currency transaction loss | (143,808 | ) | (690,748 | ) | ||||
| TOTAL OTHER EXPENSE, NET | (1,381,191 | ) | (3,259,059 | ) | ||||
| LOSS BEFORE TAXES | (4,875,451 | ) | (5,873,806 | ) | ||||
| TAX BENEFIT | 134,728 | 115,437 | ||||||
| NET LOSS | $ | (4,740,723 | ) | $ | (5,758,369 | ) | ||
| BASIC AND DILUTED NET LOSS PER SHARE | $ | (0.19 | ) | $ | (10.97 | ) | ||
| BASIC AND DILUTED WEIGHTED AVERAGE SHARES OUTSTANDING | 24,633,637 | 524,939 | ||||||
| NET LOSS | $ | (4,740,723 | ) | $ | (5,758,369 | ) | ||
| OTHER COMPREHENSIVE INCOME | ||||||||
| Unrealized foreign currency translation gain | 200,673 | 709,069 | ||||||
| TOTAL OTHER COMPREHENSIVE INCOME | 200,673 | 709,069 | ||||||
| TOTAL COMPREHENSIVE INCOME (LOSS) | $ | (4,540,050 | ) | $ | (5,049,300 | ) | ||
The accompanying notes are an integral part of these consolidated financial statements.
| F-4 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ DEFICIT FOR THE YEARS ENDED JUNE 30, 2020 AND 2019
| Preferred stock | Accumulated | |||||||||||||||||||||||||||||||||||||||||||
| Series A | Series B | Common Stock | Additional | Other | Total | |||||||||||||||||||||||||||||||||||||||
| No. of Shares | Value | No. of Shares | Value | No. of Shares | Value | Paid-in Capital | Accumulated Deficit | Treasury Stock | Comprehensive Income | Stockholders’ Deficit | ||||||||||||||||||||||||||||||||||
| Balance at June 30, 2018 | 500,000 | $ | 5,000 | 1 | $ | - | 92,859 | $ | 93 | $ | 38,214,213 | $ | (45,282,678 | ) | $ | (46,477 | ) | $ | 357,929 | $ | (6,751,920 | ) | ||||||||||||||||||||||
| Issuance of common stock for conversion of convertible debt and accrued interest | - | - | - | - | 704,258 | 704 | 3,350,079 | - | - | - | 3,350,783 | |||||||||||||||||||||||||||||||||
| Issuance of common stock under put premium | - | - | - | - | 147,200 | 147 | 1,100,233 | - | - | - | 1,100,380 | |||||||||||||||||||||||||||||||||
| Reclassification of put premium upon debt conversion | - | - | - | - | - | - | 1,824,317 | - | - | - | 1,824,317 | |||||||||||||||||||||||||||||||||
| Extinguishment of derivative liability associated with convertible notes | - | - | - | - | - | - | 1,029,039 | - | - | - | 1,029,039 | |||||||||||||||||||||||||||||||||
| Issuance of common stock for exercise of warrants | - | - | - | - | 24 | - | 30 | - | - | - | 30 | |||||||||||||||||||||||||||||||||
| Issuance of common stock for offering costs | - | - | - | - | 7,701 | 8 | 298,914 | - | - | - | 298,922 | |||||||||||||||||||||||||||||||||
| Issuance of common stock for services | - | - | - | - | 16,000 | 16 | 168,984 | - | - | - | 169,000 | |||||||||||||||||||||||||||||||||
| Stock based compensation in connection with stock option and restricted stock unit grants | - | - | - | - | - | - | 41,436 | - | - | - | 41,436 | |||||||||||||||||||||||||||||||||
| Amortization of offering costs | - | - | - | - | - | - | (313,923 | ) | - | - | - | (313,923 | ) | |||||||||||||||||||||||||||||||
| Foreign currency translation loss | - | - | - | - | - | - | - | - | - | 709,069 | 709,069 | |||||||||||||||||||||||||||||||||
| Net loss for the fiscal year ended June 30, 2019 | - | - | - | - | - | - | - | (5,758,369 | ) | - | - | (5,758,369 | ) | |||||||||||||||||||||||||||||||
| Balance at June 30, 2019 | 500,000 | 5,000 | 1 | - | 968,042 | 968 | 45,713,322 | (51,041,047 | ) | (46,477 | ) | 1,066,998 | (4,301,236 | ) | ||||||||||||||||||||||||||||||
| Issuance of units for cash | - | - | - | - | 804,518 | 804 | 424,186 | - | - | - | 424,990 | |||||||||||||||||||||||||||||||||
| Issuance of common stock for conversion of convertible debt and accrued interest | - | - | - | - | 247,619,247 | 247,619 | 1,877,555 | - | - | - | 2,125,174 | |||||||||||||||||||||||||||||||||
| Reclassification of put premium upon debt conversion | - | - | - | - | - | - | 874,924 | - | - | - | 874,924 | |||||||||||||||||||||||||||||||||
| Issuance of common stock for services | - | - | - | - | 8,728,574 | 8,729 | 104,913 | - | - | - | 113,642 | |||||||||||||||||||||||||||||||||
| Relative fair value of warrants issued with convertible debt | - | - | - | - | - | - | 375,905 | - | - | - | 375,905 | |||||||||||||||||||||||||||||||||
| Stock based compensation in connection with stock option grants and restricted stock unit grants | - | - | - | - | - | - | 300,416 | - | - | - | 300,416 | |||||||||||||||||||||||||||||||||
| Stock based compensation in connection with fair value of warrants issued for services | - | - | - | - | - | - | 984,810 | - | - | - | 984,810 | |||||||||||||||||||||||||||||||||
| Foreign currency translation gain | - | - | - | - | - | - | - | - | - | 200,673 | 200,673 | |||||||||||||||||||||||||||||||||
| Net loss for the fiscal year ended June 30, 2020 | - | - | - | - | - | - | - | (4,740,723 | ) | - | - | (4,740,723 | ) | |||||||||||||||||||||||||||||||
| Balance at June 30, 2020 | 500,000 | $ | 5,000 | 1 | $ | - | 258,120,381 | $ | 258,120 | $ | 50,656,031 | $ | (55,781,770 | ) | $ | (46,477 | ) | $ | 1,267,671 | $ | (3,641,425 | ) | ||||||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-5 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CASH FLOWS
| Years Ended June 30, | ||||||||
| 2020 | 2019 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss | $ | (4,740,723 | ) | $ | (5,758,369 | ) | ||
| Adjustments to Reconcile Net Loss to Net Cash Used in Operating Activities: | ||||||||
| Issuance and amortization of common stock and warrants for services | 1,098,452 | 245,145 | ||||||
| (Gain) loss on settlements, net | - | (14,101 | ) | |||||
| Foreign currency transaction loss (gain) | 143,808 | 690,748 | ||||||
| Depreciation expense | 2,473 | 2,306 | ||||||
| Amortization of debt discounts | 734,130 | 389,673 | ||||||
| Derivative income (expense) | (385,293 | ) | 2,472,146 | |||||
| Gain on extinguishment of debt | (67,123 | ) | (1,296,376 | ) | ||||
| Stock option and restricted stock expense | 300,416 | - | ||||||
| Non-cash interest expense | 15,000 | - | ||||||
| Put premiums on convertible debt issued | 836,724 | 706,154 | ||||||
| Changes in Assets and Liabilities: | ||||||||
| GST receivable | 3,332 | 489 | ||||||
| Prepaid expenses and other assets | 83,157 | (83,296 | ) | |||||
| Accounts payable | (59,737 | ) | (164,926 | ) | ||||
| Deferred rent | 3,394 | - | ||||||
| Employee benefit liability | 35,724 | 188,326 | ||||||
| Put premiums on convertible debt issued | (9,740 | ) | 377,846 | |||||
| Accrued interest | 156,417 | 184,198 | ||||||
| NET CASH USED IN OPERATING ACTIVITIES | (1,849,589 | ) | (2,060,037 | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||
| Purchase of equipment | - | (2,874 | ) | |||||
| NET CASH USED IN INVESTING ACTIVITIES | - | (2,874 | ) | |||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Proceeds from convertible promissory notes | 1,591,250 | 1,369,000 | ||||||
| Repayments of convertible promissory notes | - | (272,000 | ) | |||||
| Proceeds from the sale of common stock | 450,000 | 1,100,380 | ||||||
| Fees associated with offering costs | (25,010 | ) | (15,000 | ) | ||||
| Debt issuance costs | (126,000 | ) | (63,850 | ) | ||||
| Proceeds from the exercise of warrants | - | 30 | ||||||
| NET CASH PROVIDED BY FINANCING ACTIVITIES | 1,890,240 | 2,118,560 | ||||||
| Effect of exchange rate changes on cash | 23,962 | (73,176 | ) | |||||
| NET INCREASE (DECREASE) IN CASH | 64,613 | (17,527 | ) | |||||
| CASH AT BEGINNING OF YEAR | 2,394 | 19,921 | ||||||
| CASH AT END OF YEAR | $ | 67,007 | $ | 2,394 | ||||
| Supplemental Disclosure of Cash Flow Information | ||||||||
| Cash paid during the year: | ||||||||
| Interest | $ | 6,110 | $ | 100,719 | ||||
| Income Tax | $ | - | $ | - | ||||
| Supplemental Disclosure of Non-Cash Investing and Financing Activities | ||||||||
| Reduction of put premium related to conversions of convertible notes | $ | 874,924 | $ | 1,824,317 | ||||
| Conversion of convertible notes and accrued interest to common stock | $ | 1,814,336 | $ | 3,350,783 | ||||
| Discounts related to warrants issued with convertible notes | $ | 375,905 | $ | - | ||||
| Deferred financing costs associated with equity purchase agreement | $ | - | $ | 318,059 | ||||
| Discounts related to derivative liability | $ | 227,000 | $ | 180,000 | ||||
| Operating lease right-of-use asset and operating lease liability recorded on adoption of ASC 842 | $ | 48,662 | $ | - | ||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-6 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
NOTE 1 – NATURE OF OPERATIONS AND SUMMARY OF SIGNIFICANT ACCOUNTING AND REPORTING POLICIES
Nature of Operations
Propanc Biopharma, Inc. (the “Company,” “we,” “us” or “our”) was originally incorporated in Melbourne, Victoria Australia on October 15, 2007 as Propanc PTY LTD, and continues to be based in Camberwell, Victoria Australia. Since its inception, substantially all of the operations of the Company have been focused on the development of new cancer treatments targeting high-risk patients, particularly cancer survivors, who need a follow-up, non-toxic, long-term therapy designed to prevent the cancer from returning and spreading. The Company anticipates establishing global markets for its technologies. Our lead product candidate, which we refer to as PRP, is an enhanced pro-enzyme formulation designed to enhance the anti-cancer effects of multiple enzymes acting synergistically. It is currently in the preclinical phase of development.
On November 23, 2010, the Company was incorporated in the state of Delaware as Propanc Health Group Corporation. In January 2011, to reorganize the Company, we acquired all of the outstanding shares of Propanc PTY LTD on a one-for-one basis making it a wholly-owned subsidiary of the Company.
On July 22, 2016, the Company formed a wholly owned subsidiary, Propanc (UK) Limited under the laws of England and Wales for the purpose of submitting an orphan drug application to the European Medicines Agency as a small and medium-sized enterprise. As of June 30, 2020, there has been no activity within this entity.
Effective April 20, 2017, the Company changed its name to “Propanc Biopharma, Inc.” to better reflect the Company’s stage of operations and development.
The Company has filed multiple patent applications relating to its lead product, PRP. The first application was filed in October 2010. This patent has been granted and remains in force in the United States, Belgium, Czech Republic, Denmark, France, Germany, Ireland, Italy, Netherlands, Portugal, Spain, Sweden, Switzerland, Liechtenstein, Turkey, United Kingdom, Australia, China, Hong Kong, Japan, Indonesia, Israel, New Zealand, Singapore, Malaysia, South Africa, Mexico, Republic of Korea and India. In Brazil and Canada, the patent application remains under examination.
In 2016 and 2017 we filed other patent applications. Three applications were filed under the Patent Cooperation Treaty (the “PCT”). The PCT assists applicants in seeking patent protection by filing one international patent application under the PCT, which allows the applicants to seek protection for an invention in over 150 countries. Once national or regional applications are filed, the application is placed under the control of the national or regional patent offices, as applicable, in what is called the national or regional phase. One PCT application, filed in November 2016, entered the national phase in July 2018. A second application filed in January 2017 entered the national phase commencing July 2018. A third application entered the national phase in October 2018.
The Company hopes to capture and protect additional patentable subject matter based on the Company’s field of technology relating to pharmaceutical compositions of proenzymes for treating cancer by filing additional patent applications as it advances its lead product candidate, PRP, through various stages of development.
Changes in Authorized Common Stock and Reverse Split
On June 24, 2019, the Company effected a one-for-five hundred (1:500) reverse stock split whereby the Company (i) decreased the number of authorized shares of common stock, $0.001 par value per share, to 100,000,000 and (ii) decreased by a ratio of one-for-five hundred (1:500) the number of retroactively issued and outstanding shares of common stock. Proportional adjustments for the reverse stock split were made to the Company’s outstanding stock options, warrants and equity incentive plans. All share and per-share data and amounts have been retroactively adjusted as of the earliest period presented in the consolidated financial statements to reflect the reverse stock split.
On February 4, 2020 the Directors resolved to increase the Common Stock of the Company from 100,000,000 authorized shares to 1,000,000,000 authorized shares and believes that such number of authorized shares of Common Stock will be in the best interests of the Corporation and its stockholders because the Board believes that the availability of more shares of Common Stock for issuance will allow the Corporation greater flexibility in pursuing financing from investors, meeting business needs as they arise, taking advantage of favorable opportunities and responding to a changing corporate environment. The Company filed the necessary documents with the U.S. Securities and Exchange Commission on February 6, 2020 and at the date of this filing the increase in authorized shares to 1,000,000,000 has been effected on March 13, 2020.
| F-7 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Principles of Consolidation
The consolidated financial statements include the accounts of Propanc Biopharma, Inc., the parent entity, and its wholly-owned subsidiary, Propanc PTY LTD. All inter-company balances and transactions have been eliminated in consolidation. Propanc (UK) Limited was an inactive subsidiary from inception through June 30, 2020.
Use of Estimates
The preparation of financial statements in conformity with the accounting principles generally accepted in the United States of America (“US GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from these estimates. Significant estimates in the accompanying consolidated financial statements include the estimates of useful lives for depreciation, valuation of the operating lease liability and related right-of-use asset, valuation of derivatives, valuation of beneficial conversion features on convertible debt, allowance for uncollectable receivables, valuation of equity based instruments issued for other than cash, the valuation allowance on deferred tax assets and foreign currency translation due to certain average exchange rates applied in lieu of spot rates on transaction dates.
Foreign Currency Translation and Other Comprehensive Income (Loss)
The Company’s wholly owned subsidiary’s functional currency is the Australian dollar (AUD). For financial reporting purposes, the Australian dollar has been translated into the Company’s reporting currency which is the United States dollar ($) and/or (USD). Assets and liabilities are translated at the exchange rate in effect at the balance sheet date. Revenues and expenses are translated at the average rate of exchange prevailing during the reporting period. Equity transactions are translated at each historical transaction date spot rate. Translation adjustments arising from the use of different exchange rates from period to period are included as a component of stockholders’ equity (deficit) as “Accumulated other comprehensive income (loss).” Gains and losses resulting from foreign currency transactions are included in the statements of operations and comprehensive income (loss) as other comprehensive income (loss). There have been no significant fluctuations in the exchange rate for the conversion of Australian dollars to USD after the balance sheet date.
Other Comprehensive Income (Loss) for all periods presented includes only foreign currency translation gains (losses).
Assets and liabilities denominated in foreign currencies are translated into the functional currency at the exchange rates prevailing at the consolidated balance sheet date with any transaction gains and losses that arise from exchange rate fluctuations on transactions denominated in a currency other than the functional currency included in the consolidated results of operations as incurred. For the year ended June 30, 2020, the Company recognized an exchange loss of approximately $133,000 on intercompany loans made by the parent to the subsidiary which have not been repaid as at June 30, 2020.
As of June 30, 2020 and 2019, the exchange rates used to translate amounts in Australian dollars into USD for the purposes of preparing the consolidated financial statements were as follows:
| June 30, 2020 | June 30, 2019 | |||||||
| Exchange rate on balance sheet dates | ||||||||
| USD : AUD exchange rate | 0.6891 | 0.7153 | ||||||
| Average exchange rate for the period | ||||||||
| USD : AUD exchange rate | 0.6742 | 0.7009 | ||||||
Change in Accumulated Other Comprehensive Income (Loss) by component during the years ended June 30, 2020 and 2019 was as follows:
| Foreign
Currency Items: | ||||
| Beginning balance, June 30, 2018 | $ | 357,929 | ||
| Foreign currency translation gain | 709,069 | |||
| Balance, June 30, 2019 | 1,066,998 | |||
| Foreign currency translation gain | 200,673 | |||
| Ending balance, June 30, 2020 | $ | 1,267,671 | ||
| F-8 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Fair Value of Financial Instruments and Fair Value Measurements
The Company measures its financial assets and liabilities in accordance with US GAAP. For certain financial instruments, including cash and cash equivalents, accounts receivable, accounts payable and accrued liabilities, the carrying amounts approximate fair value due to their short maturities. Amounts recorded for notes payable, net of discount, and loans payable also approximate fair value because current interest rates available for debt with similar terms and maturities are substantially the same.
The Company follows accounting guidance for financial assets and liabilities. This standard defines fair value, provides guidance for measuring fair value and requires certain disclosures. This standard does not require any new fair value measurements, but rather applies to all other accounting pronouncements that require or permit fair value measurements. This guidance does not apply to measurements related to share-based payments. This guidance discusses valuation techniques, such as the market approach (comparable market prices), the income approach (present value of future income or cash flow), and the cost approach (cost to replace the service capacity of an asset or replacement cost).
The guidance utilizes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three broad levels. The following is a brief description of those three levels:
Level 1: Observable inputs such as quoted prices (unadjusted) in active markets for identical assets or liabilities.
Level 2: Inputs, other than quoted prices that are observable, either directly or indirectly. These include quoted prices for similar assets or liabilities in active markets and quoted prices for identical or similar assets or liabilities in markets that are not active.
Level 3: Unobservable inputs in which little or no market data exists, therefore developed using estimates and assumptions developed by us, which reflect those that a market participant would use.
Also see Note 12 - Derivative Financial Instruments and Fair Value Measurements.
Cash and Cash Equivalents
Cash and cash equivalents include cash on hand and at banks, short-term deposits with an original maturity of three months or less with financial institutions, and bank overdrafts. Bank overdrafts are reflected as a current liability on the balance sheets. There were no cash equivalents as of June 30, 2020 or June 30, 2019.
Property and Equipment
Property and equipment are stated at cost, net of accumulated depreciation. Expenditures for maintenance and repairs are expensed as incurred; additions, renewals, and betterments are capitalized. When property and equipment are retired or otherwise disposed of, the related cost and accumulated depreciation are removed from the respective accounts, and any gain or loss is included in operations. Depreciation of property and equipment is provided using the declining balance method. The depreciable amount is the cost less its residual value.
The estimated useful lives are as follows:
| Machinery and equipment | - 5 years |
| Furniture | - 7 years |
Patents
Patents are stated at cost and reclassified to intangible assets and amortized on a straight-line basis over the estimated future periods if and once the patent has been granted by a regulatory agency. However, the Company will expense any patent costs as long as we are in the startup stage. Accordingly, as the Company’s products are not currently approved for market, all patent costs incurred from 2013 through June 30, 2020 were expensed immediately. This practice of expensing patent costs immediately ends when a product receives market authorization from a government regulatory agency.
Impairment of Long-Lived Assets
In accordance with ASC 360-10, “Long-lived assets,” which include property and equipment and intangible assets, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of long-lived assets to be held and used is measured by a comparison of the carrying amount of an asset to the estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated undiscounted future cash flows, an impairment charge is recognized by the amount by which the carrying amount of the asset exceeds the fair value of the assets. Fair value is generally determined using the asset’s expected future discounted cash flows or market value, if readily determinable.
| F-9 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Employee Benefit/Liability
Liabilities arising in respect of wages and salaries, accumulated annual leave, accumulated long service leave and any other employee benefits expected to be settled within twelve months of the reporting date are measured based on the employees remuneration rates applicable at the reporting date. All other employee benefit liabilities are measured at the present value of the estimated future cash outflow to be made in respect of services provided by employees up to the reporting date. All employee liabilities are owed within the next twelve months.
Australian Goods and Services Tax (“GST”)
Revenues, expenses and balance sheet items are recognized net of the amount of GST, except payable and receivable balances which are shown inclusive of GST. The GST incurred is payable on revenues to, and recoverable on purchases from, the Australian Taxation Office.
Cash flows are presented in the statements of cash flow on a gross basis, except for the GST component of investing and financing activities, which are disclosed as operating cash flows.
As of June 30, 2020 and 2019, the Company was owed $2,015 and $5,439, respectively, from the Australian Taxation Office. These amounts were fully collected subsequent to the balance sheet reporting dates.
Derivative Instruments
ASC Topic 815, Derivatives and Hedging (“ASC Topic 815”), establishes accounting and reporting standards for derivative instruments and for hedging activities by requiring that all derivatives be recognized in the balance sheet and measured at fair value. Gains or losses resulting from changes in the fair value of derivatives are recognized in earnings. On the date of conversion or payoff of debt, the Company records the fair value of the conversion shares, removes the fair value of the related derivative liability, removes any discounts and records a net gain or loss on debt extinguishment. On July 1, 2019 the Company adopted ASU 2017-11 under which down-round Features in Financial Instruments will no longer cause derivative treatment. The Company applies the modified prospective method of adoption. There were no cumulative effects on adoption.
Convertible Notes With Variable Conversion Options
The Company has entered into convertible notes, some of which contain variable conversion options, whereby the outstanding principal and accrued interest may be converted, by the holder, into common shares at a fixed discount to the price of the common stock at or around the time of conversion. The Company treats these convertible notes as stock settled debt under ASC 480, “Distinguishing Liabilities from Equity” and measures the fair value of the notes at the time of issuance, which is the result of the share price discount at the time of conversion and records the put premium as interest expense.
Income Taxes
The Company is governed by Australia and United States income tax laws, which are administered by the Australian Taxation Office and the United States Internal Revenue Service, respectively. The Company follows ASC 740 “Accounting for Income Taxes,” when accounting for income taxes, which requires an asset and liability approach to financial accounting and reporting for income taxes. Deferred income tax assets and liabilities are computed annually for temporary differences between the financial statements and tax bases of assets and liabilities that will result in taxable or deductible amounts in the future based on enacted tax laws and rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized. Income tax expense is the tax payable or refundable for the period plus or minus the change during the period in deferred tax assets and liabilities.
The Company follows ASC 740, Sections 25 through 60, “Accounting for Uncertainty in Income Taxes.” These sections provide detailed guidance for the financial statement recognition, measurement and disclosure of uncertain tax positions recognized in the financial statements. Tax positions must meet a “more-likely-than-not” recognition threshold at the effective date to be recognized upon the adoption of ASC 740 and in subsequent periods.
| F-10 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Research and Development Costs and Tax Credits
In accordance with ASC 730-10, “Research and Development-Overall,” research and development costs are expensed when incurred. Total research and development costs for the fiscal years ended June 30, 2020 and 2019 were $179,987 and $260,335, respectively.
The Company may apply for research and development tax concessions with the Australian Taxation Office on an annual basis. Although the amount is possible to estimate at year end, the Australian Taxation Office may reject or materially alter the claim amount. Accordingly, the Company does not recognize the benefit of the claim amount until cash receipt since collectability is not certain until such time. The tax concession is a refundable credit. If the Company has net income, then the Company can receive the credit which reduces its income tax liability. If the Company has net losses, then the Company may still receive a cash payment for the credit, however, the Company’s net operating loss carryforwards are reduced by the gross equivalent loss that would produce the credit amount when the income tax rate is applied to that gross amount. The concession is recognized as an income tax benefit, in operations, upon receipt.
During each of the fiscal years ended June 30, 2020 and 2019, the Company applied for, and received from the Australian Taxation Office, a research and development tax credit in the amount of $134,728 and $115,437, respectively, which is reflected as a tax benefit in the accompanying consolidated statements of operations and comprehensive income (loss).
Stock Based Compensation
The Company records stock-based compensation in accordance with ASC 718, “Stock Compensation”. ASC 718 requires the fair value of all stock-based employee compensation awarded to employees to be recorded as an expense over the shorter of the service period or the vesting period. The Company values employee and non-employee stock-based compensation at fair value using the Black-Scholes Option Pricing Model.
The Company adopted ASU 2018-07 and accounts for non-employee share-based awards in accordance with the measurement and recognition criteria of ASC 718 and recognizes the fair value of such awards over the service period. The Company used the modified prospective method of adoption. There was no cumulative effect of adoption on July 1, 2019.
Revenue Recognition
The Company adopted and implemented on July 1, 2018, ASC 606 – Revenue from Contracts with Customers (“ASC 606”). ASC 606 did not have a material impact on the consolidated financial statements.
Upon implementation of ASC 606, the Company recognizes revenue in accordance with that core principle by applying the following steps:
Step 1: Identify the contract(s) with a customer.
Step 2: Identify the performance obligations in the contract.
Step 3: Determine the transaction price.
Step 4: Allocate the transaction price to the performance obligations in the contract.
Step 5: Recognize revenue when (or as) the entity satisfies a performance obligation.
Subject to these criteria, the Company intends to recognize revenue relating to royalties on product sales in the period in which the sale occurs and the royalty term has begun.
Legal Expenses
All legal costs for litigation are charged to expense as incurred.
Leases
In February 2016, the Financial Accounting Standards Board (“FASB”) issued ASU 2016-02, Leases (Topic 842). The updated guidance requires lessees to recognize lease assets and lease liabilities for most operating leases. In addition, the updated guidance requires that lessors separate lease and non-lease components in a contract in accordance with the new revenue guidance in ASC 606. This guidance is effective for interim and annual reporting periods beginning after December 15, 2018. The Company adopted this guidance effective July 1, 2019.
On July 1, 2019, the Company adopted ASU No. 2016-02, applying the package of practical expedients to leases that commenced before the effective date whereby the Company elected to not reassess the following: (i) whether any expired or existing contracts contain leases and; (ii) initial direct costs for any existing leases. For contracts entered into on or after the effective date, at the inception of a contract the Company assessed whether the contract is, or contains, a lease. The Company’s assessment is based on: (1) whether the contract involves the use of a distinct identified asset, (2) whether we obtain the right to substantially all the economic benefit from the use of the asset throughout the period, and (3) whether it has the right to direct the use of the asset. The Company will allocate the consideration in the contract to each lease component based on its relative stand-alone price to determine the lease payments.
| F-11 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Operating lease ROU assets represents the right to use the leased asset for the lease term and operating lease liabilities are recognized based on the present value of future minimum lease payments over the lease term at commencement date. As most leases do not provide an implicit rate, the Company use an incremental borrowing rate based on the information available at the adoption date in determining the present value of future payments. Lease expense for minimum lease payments is amortized on a straight-line basis over the lease term and is included in general and administrative expenses in the condensed consolidated statements of operations.
Basic and Diluted Net Loss Per Common Share
Basic net loss per share is computed by dividing the net loss by the weighted average number of common shares outstanding during the period. Diluted net loss per common share is computed by dividing the net loss by the weighted average number of common shares outstanding for the period and, if dilutive, potential common shares outstanding during the period. Potentially dilutive securities consist of the incremental common shares issuable upon exercise of common stock equivalents such as stock options, warrants and convertible debt instruments. Potentially dilutive securities are excluded from the computation if their effect is anti-dilutive. As a result, the basic and diluted per share amounts for all periods presented are identical. As of June 30, 2019, there were 59 warrants outstanding, 59,644 stock options and 10 convertible notes payable, which notes are convertible into approximately 1,810,347 shares of the Company’s common stock. As of June 30, 2020, there were 151,170,514 warrants outstanding, 59,644 stock options and 11 convertible notes payable, which notes are convertible into approximately 439,113,281 shares of the Company’s common stock. Each holder of the notes has agreed to a 4.99% beneficial ownership conversion limitation (subject to certain noteholders’ ability to increase such limitation to 9.99% upon 60 days’ notice to the Company), and each note may not be converted during the first six-month period from the date of issuance. Such securities are considered dilutive securities which were excluded from the computation since the effect is anti-dilutive.
Recent Accounting Pronouncements
We have reviewed the FASB issued ASU accounting pronouncements and interpretations thereof that have effectiveness dates during the periods reported and in future periods. We have carefully considered the new pronouncements that alter previous generally accepted accounting principles and do not believe that any new or modified principles will have a material impact on the Company’s reported financial position or operations in the near term. The applicability of any standard is subject to the formal review of the Company’s financial management.
NOTE 2 – GOING CONCERN
The accompanying consolidated financial statements have been prepared in conformity with US GAAP, which contemplate continuation of the Company as a going concern. For the fiscal year ended June 30, 2020, the Company had no revenues, had a net loss of $4,740,723 and had net cash used in operations of $1,849,589. Additionally, as of June 30, 2020, the Company had a working capital deficit, stockholders’ deficit and accumulated deficit of $3,670,921, $3,641,425 and $55,781,770, respectively. It is management’s opinion that these conditions raise substantial doubt about the Company’s ability to continue as a going concern for a period of at least twelve months from the date of this filing.
The consolidated financial statements do not include any adjustments to reflect the possible future effect on the recoverability and classification of assets or the amounts and classifications of liabilities that may result from the outcome of this uncertainty.
Successful completion of the Company’s development program and, ultimately, the attainment of profitable operations are dependent upon future events, including obtaining adequate financing to fulfill its development activities, acceptance of the Company’s patent applications, obtaining additional sources of suitable and adequate financing and ultimately achieving a level of sales adequate to support the Company’s cost structure and business plan. The Company’s ability to continue as a going concern is also dependent on its ability to further develop and execute on its business plan. However, there can be no assurances that any or all of these endeavors will be successful.
In March 2020, the outbreak of COVID-19 (coronavirus) caused by a novel strain of the coronavirus was recognized as a pandemic by the World Health Organization, and the outbreak has become increasingly widespread in the United States, Europe and Australia, including in each of the areas in which the Company operates. The COVID-19 (coronavirus) outbreak has had a notable impact on general economic conditions, including but not limited to the temporary closures of many businesses, “shelter in place” and other governmental regulations, reduced business and consumer spending due to both job losses, reduced investing activity and M&A transactions, among many other effects attributable to the COVID-19 (coronavirus), and there continue to be many unknowns. While to date the Company has not been required to stop operating, management is evaluating its use of its office space, virtual meetings and the like. The Company continues to monitor the impact of the COVID-19 (coronavirus) outbreak closely. The extent to which the COVID-19 (coronavirus) outbreak will impact our operations, ability to obtain financing or future financial results is uncertain.
| F-12 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
NOTE 3 – PROPERTY AND EQUIPMENT
Property and equipment consist of the following as of June 30,
| 2020 | 2019 | |||||||
| Office equipment at cost | $ | 26,299 | $ | 26,749 | ||||
| Less: Accumulated depreciation | (20,552 | ) | (18,332 | ) | ||||
| Total property, plant, and equipment | $ | 5,747 | $ | 8,417 | ||||
Depreciation expense for the years ended June 30, 2020 and 2019 were $2,473 and $2,306, respectively
NOTE 4 – DUE TO FORMER DIRECTOR - RELATED PARTY
Due to director - related party represents unsecured advances made primarily by a former director for operating expenses on behalf of the Company such as intellectual property and formation expenses. The expenses were paid for on behalf of the Company and are due upon demand. The Company is currently not being charged interest under these advances. The total amount owed the former director at June 30, 2020 and 2019 is $30,639 and $31,164, respectively. The Company plans to repay the advances as its cash resources allow. (see Note 10)
NOTE 5 – LOANS AND NOTES PAYABLE
Loans from Directors and Officer - Related Parties
Loans from the Company’s directors and officer at June 30, 2020 and 2019 were $50,993 and $51,867, respectively. The loans bear no interest and are all payable on demand. The Company did not repay any amount on these loans during the years ended June 30, 2020 and 2019, respectively. (See Note 10)
NOTE 6 – CONVERTIBLE NOTES
The Company’s convertible notes outstanding at June 30, 2020 and 2019 were as follows:
| June 30, 2020 | June 30, 2019 | |||||||
| Convertible notes and debenture | $ | 1,029,496 | $ | 1,076,785 | ||||
| Unamortized discounts | (126,667 | ) | (131,893 | ) | ||||
| Accrued interest | 80,101 | 99,482 | ||||||
| Premium, net | 574,804 | 613,003 | ||||||
| Convertible notes, net | $ | 1,557,734 | $ | 1,657,377 | ||||
Eagle Equities Financing Agreements
December 12, 2016 Securities Purchase Agreement
On December 12, 2016, the Company entered into a Securities Purchase Agreement, with Eagle Equities, pursuant to which Eagle Equities purchased two 8% convertible redeemable junior subordinated promissory notes, each in the principal amount of $100,000. The first note (the “December 12 Note”) was funded with cash and the second note (the “December 12 Eagle Back-End Note”) was initially paid for by an offsetting promissory note issued by Eagle Equities to the Company (the “December 12 Note Receivable”). The terms of the December 12 Eagle Back-End Note require cash funding prior to any conversion thereunder. The December 12 Note Receivable was due on December 12, 2017, unless certain conditions were not met, in which case both the December 12 Eagle Back-End Note and the December 12 Note Receivable may both be cancelled. Both the December 12 Note and the December 12 Eagle Back-End Note had a maturity date one year from the date of issuance upon which any outstanding principal and interest is due and payable. The outstanding principal amounts plus accrued interest under both the December 12 Note and the December 12 Eagle Back-End Note were convertible into the Company’s common stock at a conversion price equal to 60% of the lowest closing bid price of the common stock for the ten trading days prior to the conversion, subject to adjustment in certain events. On April 11, 2017, the Company received payment of the December 12 Note Receivable in the amount of $100,000 that offset the December Eagle Back-End Note. Proceeds from the Note Receivable of $5,000 were paid directly to legal fees resulting in net cash proceeds of $95,000 received by the Company. As a result, the December 12 Eagle Back-End Note then became convertible. The December 12 Note and the December 12 Eagle Back-End Note were treated as stock settled debt under ASC 480 and accordingly the Company recorded a put premium of $66,667 as each of the notes were funded. As of June 30, 2018, the outstanding principal under the December 12 Note along with $8,296 of accrued interest was fully converted into shares of the Company’s common stock. As of June 30, 2019, the outstanding balance of $100,000 under the December 12 Eagle Back-End Note along with $13,144 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. There was no outstanding balance as of June 30, 2019.
| F-13 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
January 27, 2017 Securities Purchase Agreement
On January 27, 2017, the Company entered into a Securities Purchase Agreement with Eagle Equities, LLC, pursuant to which Eagle Equities purchased two 8% convertible redeemable junior subordinated promissory notes, each in the principal amount of $230,000. The first note (the “January 2017 Eagle Note”) was funded with cash and the second note (the “January 2017 Eagle Back-End Note”) was initially paid for by an offsetting promissory note issued by Eagle Equities to the Company (the “January 2017 Eagle Note Receivable”). The terms of the January 2017 Eagle Back-End Note require cash funding prior to any conversion thereunder. The January 2017 Eagle Note Receivable was due September 27, 2017, unless certain conditions were not met, in which case both the January 2017 Eagle Back-End Note and the January 2017 Eagle Note Receivable may both be cancelled. Both the January 2017 Eagle Note and the January 2017 Eagle Back-End Note had a maturity date one year from the date of issuance upon which any outstanding principal and interest is due and payable. The outstanding principal amounts plus accrued interest under both the January 2017 Eagle Note and the January 2017 Eagle Back-End Note are convertible into common stock of the Company at a conversion price equal to 60% of the lowest closing bid price of the common stock for the ten trading days prior to the conversion, subject to adjustment in certain events. On May 4, 2017, the Company received a partial payment of the January 2017 Note Receivable in the amount of $40,000 and on June 3, 2017 the balance of $190,000 was funded, of which $11,250 was paid directly to legal fees. As a result, the January 2017 Eagle Back-End Note then became convertible. The January 2017 Eagle Note and the January 2017 Eagle Back-End Note were treated as stock settled debt under ASC 480 and accordingly the Company is recording a put premium of $153,333 as each of the notes were funded. As of June 30, 2018, the outstanding principal under the January 2017 Eagle Note along with $14,988 of accrued interest was fully converted. As of June 30, 2019, the outstanding balance of $230,000 under the January 2017 Eagle Back-End Note along with $33,356 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. There was no outstanding balance as of June 30, 2019.
March 1, 2017 Securities Purchase Agreement
On March 1, 2017, the Company entered into a Securities Purchase Agreement with Eagle Equities, pursuant to which Eagle Equities purchased two 8% convertible redeemable junior subordinated promissory notes, each in the principal amount of $220,500. The first note (the “March 2017 Eagle Note”) was funded with cash and the second note (the “March 2017 Eagle Back-End Note”) was initially paid for by an offsetting promissory note issued by Eagle Equities to the Company (the “March 2017 Eagle Note Receivable”). The terms of the March 2017 Eagle Back-End Note require cash funding prior to any conversion thereunder. Both the March 2017 Eagle Note and the March 2017 Eagle Back-End Note had a maturity date of March 1, 2018, upon which any outstanding principal and interest was due and payable. The outstanding principal amounts plus accrued interest under both the March 2017 Eagle Note and the March 2017 Eagle Back-End Note were convertible into shares of common stock, of the Company at a conversion price equal to 60% of the lowest closing bid price of the common stock for the ten trading days prior to the conversion, subject to adjustment in certain events. On July 5, 2017, the Company received payment of the March 2017 Eagle Note Receivable in the amount of $220,500 that offset the March 2017 Eagle Back-End Note. Proceeds from the March 2017 Eagle Note Receivable of $10,500 were paid directly to legal fees resulting in net cash proceeds of $210,000 received by the Company. As a result, the March 2017 Eagle Back-End Note then became convertible. The March 2017 Eagle Note and the March 2017 Eagle Back-End Note were treated as stock settled debt under ASC 480 and accordingly the Company recorded a put premium of $147,000 as each of the notes were funded. As of June 30, 2018, the outstanding principal balance under the March 2017 Eagle Note along with $20,061 of accrued interest was fully converted. As of June 30, 2019, the outstanding balance of $220,500 under the March 2017 Back-End Note along with $19,526 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in a full reduction of the put premium. There was no outstanding balance as of June 30, 2019.
August 9, 2017 Securities Purchase Agreement
On August 9, 2017, the Company entered into a Securities Purchase Agreement dated as of August 8, 2017, with Eagle Equities, pursuant to which Eagle Equities purchased two 8% convertible redeemable junior subordinated promissory notes, each in the principal amount of $200,000. The first note (the “August 2017 Eagle Note”) was funded with cash and the second note (the “August 2017 Eagle Back-End Note”) was initially paid for by an offsetting promissory note issued by Eagle Equities to the Company (the “August 2017 Eagle Note Receivable”). The terms of the August 2017 Eagle Back-End Note require cash funding prior to any conversion thereunder. The August 2017 Eagle Note Receivable was due August 8, 2018, unless certain conditions were not met, in which case both the August 2017 Eagle Back-End Note and the August 2017 Eagle Note Receivable may both be cancelled. Both the August 2017 Eagle Note and the August 2017 Eagle Back-End Note had a maturity date one year from the date of issuance upon which any outstanding principal and interest is due and payable. The outstanding principal amounts plus accrued interest under both the August 2017 Eagle Note and the August 2017 Eagle Back-End Note were convertible into common stock of the Company at a conversion price equal to 60% of the lowest closing bid price of the common stock for the ten trading days prior to the conversion, subject to adjustment in certain events. On September 14, 2017, the Company received payment of the August 2017 Eagle Note Receivable in the amount of $200,000 that offset the August 2017 Eagle Back-End Note. Proceeds from the August 2017 Eagle Note Receivable of $10,000 were paid directly to legal fees resulting in net cash proceeds of $190,000 received by the Company. As a result, the August 2017 Eagle Back-End Note then became convertible. The August 2017 Eagle Note and the August 2017 Eagle Back-End Note were treated as stock settled debt under ASC 480 and accordingly the Company recorded a put premium of $133,333 as each of the notes were funded. As of June 30, 2018 $120,000 of principal under the August 2017 Eagle Note along with $5,273 in interest was converted. As of June 30, 2019, the remaining outstanding balance of $80,000 under the August 2017 Eagle Note along with $6,850 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. As of June 30, 2019, the remaining outstanding principal balance of $200,000 under the August 2017 Eagle Back-Note along with $30,568 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. There was no outstanding balance as of June 30, 2019.
| F-14 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
October 25, 2017 Securities Purchase Agreement
On November 3, 2017, the Company entered into a Securities Purchase Agreement dated as of October 25, 2017, with Eagle Equities, pursuant to which Eagle Equities purchased two 8% convertible redeemable junior subordinated promissory notes, each in the principal amount of $200,000. The first note (the “October 2017 Eagle Note”) was funded with cash and the second note (the “October 2017 Eagle Back-End Note”) was initially paid for by an offsetting promissory note issued by Eagle Equities to the Company (the “October 2017 Eagle Note Receivable”). The terms of the October 2017 Eagle Back-End Note require cash funding prior to any conversion thereunder. The October 2017 Eagle Note Receivable was due June 25, 2018, unless certain conditions were not met, in which case both the October 2017 Eagle Back-End Note and the October 2017 Eagle Note Receivable may both be cancelled. Both the October 2017 Eagle Note and the October 2017 Eagle Back-End Note had a maturity date one year from the date of issuance upon which any outstanding principal and interest is due and payable. The amounts cash funded plus accrued interest under both the October 2017 Eagle Note and the October 2017 Eagle Back-End Note were convertible into common stock, par value $0.001 of the Company at a conversion price equal to 60% of the lowest closing bid price of the common stock for the ten trading days prior to the conversion, subject to adjustment in certain events. On December 6, 2017, the Company received payment of the October 2017 Eagle Note Receivable in the amount of $200,000 that offset the October 2017 Eagle Back-End Note. Proceeds from the October 2017 Eagle Note Receivable of $10,000 were paid directly to legal fees resulting in net cash proceeds of $190,000 received by the Company. As a result, the October 2017 Eagle Back-End Note then became convertible. The October 2017 Eagle Note and the October 2017 Eagle Back-End Note were treated as stock settled debt under ASC 480 and accordingly the Company recorded a put premium of $133,333 as each of the notes were funded. As of June 30, 2019, the outstanding principal balance under the October 2017 Eagle Note along with $14,653 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. There was no outstanding balance as of June 30, 2019.
December 29, 2017 Securities Purchase Agreement
The Company entered into an executory contract on December 29, 2017, whereby the Company entered into a securities purchase agreement with Eagle Equities, pursuant to which Eagle Equities purchased a convertible promissory note (the “December 2017 Eagle Note”) from the Company in the aggregate principal amount of $532,435, with principal and the interest thereon convertible into shares of the Company’s common stock at the option of Eagle Equities at any time. The transactions closed on January 2, 2018.
The December 2017 Eagle Note contained an original issue discount of $25,354 such that the purchase price was $507,081. The maturity date of the December 2017 Eagle Note was December 29, 2018. The Company was in discussions with Eagle Equities to extend the maturity date. The December 2017 Eagle Note bore interest at a rate of 8% per annum, which interest were to be paid by the Company to Eagle Equities in shares of the Company’s common stock upon receipt of a notice of conversion by the Company from Eagle Equities at any time.
The Company recorded $20,065 of accrued interest for the December 2017 Eagle Note and total principal outstanding as of June 30, 2019 under the December 2017 Eagle Note was $171,965 following conversion of $360,470 of principal and $43,535 of accrued interest during the fiscal year ended June 30, 2019. The Company recorded $0 of accrued interest for the December 2017 Eagle Note and total principal outstanding as of June 30, 2020 under the December 2017 Eagle Note was $0 following conversion of the remaining principal $171,965 and $24,751 of accrued interest during the fiscal year to June 30, 2020. Accordingly, there was no outstanding balance as of June 30, 2020.
Eagle Equities had the option to convert all or any amount of the principal face amount of the December 2017 Eagle Note, at any time, for shares of the Company’s common stock at a price equal to 60% of the lowest closing bid price of the Company’s common stock as reported on the OTCQB for the ten prior trading days, including the day upon which the Company receives a notice of conversion from Eagle Equities. The note was treated as stock settled debt under ASC 480 and accordingly the Company recorded a $354,956 put premium of which $240,313 was released to additional paid in capital following conversion of $360,470 of principal during the fiscal year to June 30, 2019, and a further $114,643 was released to additional paid in capital following conversion of the remaining principal of $171,965 during the fiscal year to June 30, 2020.
Upon an event of default, principal and accrued interest will become immediately due and payable under the notes. Additionally, upon an event of default, both notes will accrue interest at a default interest rate of 24% per annum or the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions. In April 2020, Eagle Equities agreed to waive the 24% default interest on this note.
| F-15 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
June 14, 2018 Securities Purchase Agreement
Effective June 14, 2018, the Company entered into a securities purchase agreement with Eagle Equities, pursuant to which Eagle Equities purchased a convertible promissory note (the “June 2018 Eagle Note”) from the Company in the aggregate principal amount of $105,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Eagle Equities any time after the six-month anniversary of the June 2018 Eagle Note. The transactions contemplated by the Purchase Agreement closed on June 19, 2018. Pursuant to the terms of the Purchase Agreement, Eagle Equities deducted $5,000 from the principal payment due under the June 2018 Eagle Note, at the time of closing, to be applied to its legal expenses.
The maturity date of the June 2018 Eagle Note was on June 14, 2019. The June 2018 Eagle Note bears interest at a rate of 8% per annum, which interest shall be paid by the Company to Eagle Equities in shares of the Company’s common stock upon receipt of a notice of conversion by the Company from Eagle Equities at any time after the six-month anniversary of the June 2018 Eagle Note.
Additionally, Eagle Equities had the option to convert all or any amount of the principal face amount of the June 2018 Eagle Note, at any time, for shares of the Company’s common stock at a price equal to 60% of the lowest closing bid price of the Company’s common stock as reported on the OTC quotation system for the ten prior trading days, including the day upon which the Company receives a notice of conversion from Eagle Equities. However, in the event that the Company’s common stock is restricted by the Depository Trust Company (“DTC”) for any reason, the Conversion Price shall be lowered to 50% of the lowest closing bid price for the duration of such restriction. If the Company fails to maintain a reserve of shares of its common stock at least four times the number of shares issuable upon conversion of the Note for at least 60 days after the issuance of the Note, the conversion discount shall be increased by 10%. Notwithstanding the foregoing, Eagle Equities shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Eagle Equities and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock. The June 2018 Eagle Note was treated as stock settled debt under ASC 480 and accordingly, the Company recorded a $70,000 put premium which was released to additional paid in capital upon conversion of the note as discussed above.
Upon an event of default, principal and accrued interest will become immediately payable under the note. Interest on the outstanding principal shall accrue at a default interest rate of 24% per annum or at the highest rate permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
No payments or conversions occurred in fiscal 2018.
As of June 30, 2019, the remaining outstanding principal balance of $105,000 under the June 14, 2018 Eagle Equities Note along with $6,674 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. There was no outstanding balance as of June 30, 2019.
July 13, 2018 Securities Purchase Agreement
Effective July 13, 2018, the Company entered into a securities purchase agreement with Eagle Equities, pursuant to which Eagle Equities purchased a convertible promissory note (the “July 2018 Note”) from the Company in the aggregate principal amount of $75,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Eagle Equities any time after the six month anniversary of the July 2018 Eagle Note. The transaction closed on July 16, 2018 and on July 19, 2018 the Company received proceeds of $71,250 as $3,750 was paid directly to legal fees.
The maturity date of the July 2018 Eagle Note was July 13, 2019. The July 2018 Eagle Note bears interest at a rate of 8% per annum, which interest shall be paid by the Company to Eagle Equities in shares of the Company’s common stock upon receipt of a notice of conversion by the Company from Eagle Equities at any time after the six-month anniversary of the Note.
Additionally, Eagle Equities had the option to convert all or any amount of the principal face amount of the July 2018 Eagle Note, at any time, for shares of the Company’s common stock at a price equal to 60% of the lowest closing bid price of the Company’s common stock for the ten prior trading days, including the day upon which the Company receives a notice of conversion, subject to adjustment in certain events. Eagle Equities shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Eagle Equities and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock. The July 2018 Eagle Note was treated as stock settled debt under ASC 480 and accordingly, the Company recorded a $50,000 put premium of which $50,000 was released in fiscal 2020 to additional paid in capital following the full conversion of the note.
| F-16 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
The Company recorded $5,786 of accrued interest and the total principal outstanding under the July 2018 Eagle Note was $75,000 as of June 30, 2019. The Company recorded $0 of accrued interest and the total principal outstanding under the July 2018 Eagle Note was $0 as of June 30, 2020 following conversion of $75,000 of principal and $9,300 of accrued interest during the fiscal year ended June 30, 2020. The Company had the right to prepay the July 2018 Eagle Note with certain penalties until January 9, 2019. No prepayment was made as of such date. As a result, the July 2018 Eagle Note was convertible.
Upon an event of default, principal and accrued interest will become immediately due and payable under the notes. Additionally, upon an event of default, both notes will accrue interest at a default interest rate of 24% per annum or the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions. In April 2020, Eagle Equities agreed to waive the 24% default interest on this note.
August 29, 2018 Securities Purchase Agreement
Effective August 29, 2018, the Company entered into a securities purchase agreement with Eagle Equities, pursuant to which Eagle Equities purchased a convertible promissory note (the “August 2018 Eagle Note”) from the Company in the aggregate principal amount of $105,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Eagle Equities any time after the six-month anniversary of the August 2018 Eagle Note. The transactions contemplated by the agreement closed on August 30, 2018.
The maturity date of the August 29, 2018 Eagle Note was August 29, 2019. The Company is currently in discussions with Eagle Equities to extend the maturity date. The August 2018 Eagle Note bears interest at a rate of 8% per annum, which interest shall be paid by the Company to Eagle Equities in shares of the Company’s common stock upon receipt of a notice of conversion by the Company from Eagle Equities at any time after the six-month anniversary of the August 2018 Eagle Note.
Additionally, Eagle Equities has the option to convert all or any amount of the principal face amount of the August 2018 Eagle Note, at any time, into shares of the Company’s common stock at a price equal to 60% of the lowest closing bid price (the “Closing Bid Price”) of the Company’s common stock as reported on the OTC Markets quotation system for the ten prior trading days, including the day upon which the Company receives a notice of conversion from Eagle Equities (the “Conversion Price”). However, in the event that the Company’s common stock is restricted by the DTC for any reason, the Conversion Price shall be lowered to 50% of the lowest Closing Bid Price for the duration of such restriction. If the Company fails to maintain a reserve of shares of its common stock at least four times the number of shares issuable upon conversion of the August 2018 Eagle Note for at least 60 days after the issuance of the August 28, 2018 Eagle Note, the conversion discount shall be increased by 10%. Notwithstanding the foregoing, Eagle Equities shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Eagle Equities and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock. The Company had the right to prepay the August 2018 Eagle Note with certain penalties until February 25, 2019. No prepayment was made as of such date. As a result, the August 2018 Eagle Note then became convertible. The August 2018 Eagle Note is treated as stock settled debt under ASC 480 and accordingly, the Company recorded a $70,000 put premium of which $17,000 was released to additional paid in capital following conversion of principal during the fiscal year to June 30, 2020.
The Company recorded $7,042 of accrued interest and the total principal outstanding under the August 2018 Eagle Note was $105,000 as of June 30, 2019.The Company recorded $11,663 of accrued interest and the total principal outstanding under the August 2018 Eagle Note was $79,500 as of June 30, 2020 following conversion of $25,500 of principal and $3,788 of accrued interest during the fiscal year ended June 30, 2020.
Upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 24% per annum or at the highest rate permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions. In April 2020, Eagle Equities agreed to waive the 24% default interest on this note, however, the note is currently past due.
October 2, 2018 Securities Purchase Agreement
Effective October 2, 2018, the Company entered into a securities purchase agreement with Eagle Equities, pursuant to which Eagle Equities purchased a convertible promissory note (the “October 2018 Eagle Note”) from the Company in the aggregate principal amount of $210,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Eagle Equities any time after the six-month anniversary of the October 2018 Eagle Note. The transactions contemplated by the purchase agreement closed on October 3, 2018. Pursuant to the terms of the purchase agreement, Eagle Equities deducted $10,000 from the principal payment due under the October 2018 Eagle Note, at the time of closing, to be applied to its legal expenses.
The maturity date of the October 2018 Eagle Note was October 2, 2019. The October 2018 Eagle Note shall bore interest at a rate of 8% per annum, which interest shall be paid by the Company to Eagle Equities in shares of common stock upon receipt of a notice of conversion by the Company from Eagle Equities at any time after the six-month anniversary of the October 2018 Eagle Note.
| F-17 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Additionally, Eagle Equities had the option to convert all or any amount of the principal amount of the October 2018 Eagle Note, at any time, for shares of the Company’s common stock at a price equal to 60% of the lowest closing bid price of the Company’s common stock for the ten prior trading days, including the day upon which the Company receives a notice of conversion, subject to adjustment in certain events. Eagle Equities shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Eagle Equities and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock. No prepayment was made as of such date. As a result, the October 2018 Eagle Note then became convertible. The October 2, 2018 Eagle Note was treated as stock settled debt under ASC 480 and accordingly, the Company recorded a $140,000 put premium of which $140,000 was released to additional paid in capital following conversion of principal during the year ended June 30, 2020.
The Company recorded $12,473 of accrued interest and the total principal outstanding under the October 2018 Eagle Note was $210,000 as of June 30, 2019. The Company recorded $0 of accrued interest and the total principal outstanding under the October 2018 Eagle Note was $0 as of June 30, 2020 following conversion of $210,000 of principal and $25,725 of accrued interest during the year ended June 30, 2020. The Company had the right to prepay the October 2018 Eagle Note with certain penalties until March 31, 2019.
Upon an event of default, principal and accrued interest will become immediately due and payable under the notes. Additionally, upon an event of default, both notes will accrue interest at a default interest rate of 24% per annum or the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions. In April 2020, Eagle Equities agreed to waive the 24% default interest on this note.
November 30, 2018 Securities Purchase Agreement
Effective November 30, 2018, the Company entered into a securities purchase agreement with Eagle Equities, pursuant to which Eagle Equities purchased a convertible promissory note (the “November 2018 Eagle Note”) from the Company in the aggregate principal amount of $105,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Eagle Equities any time after the six-month anniversary of the November 2018 Eagle Note. The transactions contemplated by the purchase agreement closed on December 3, 2018. Pursuant to the terms of the purchase agreement, Eagle Equities deducted $5,000 from the principal payment due under the November 2018 Eagle Note, at the time of closing, to be applied to its legal expenses.
The maturity date of the November 2018 Eagle Note was November 30, 2019. The November 2018 Eagle Note bore interest at a rate of 8% per annum, which interest shall be paid by the Company to Eagle Equities in shares of common stock upon receipt of a notice of conversion by the Company from Eagle Equities at any time after the six-month anniversary of the November 2018 Eagle Note.
Additionally, Eagle Equities had the option to convert all or any amount of the principal amount of the November 2018 Eagle Note, at any time, for shares of the Company’s common stock at a price equal to 61% of the lowest closing bid price (the “Closing Bid Price”) of the Company’s common stock as reported on the OTC Markets Group, Inc. quotation system for the ten prior trading days, including the day upon which the Company receives a notice of conversion from Eagle Equities (the “Conversion Price”). However, in the event that the Company’s common stock is restricted by the Depository Trust Company for any reason, the Conversion Price shall be lowered to 51% of the lowest Closing Bid Price for the duration of such restriction. If the Company fails to maintain a reserve of shares of its common stock at least two and a half times the number of shares issuable upon conversion of the November 2018 Eagle Note for at least 60 days after the issuance of the November 2018 Eagle Note, the conversion discount shall be increased by 10%. Notwithstanding the foregoing, Eagle Equities shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Eagle Equities and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock. The November 2018 Eagle Note was treated as stock settled debt under ASC 480 and accordingly, the Company recorded a $67,131 put premium of which $67,131 was released to additional paid in capital following conversion of principal during the year ended June 30, 2020.
The Company recorded $4,879 of accrued interest and the total principal outstanding under the November 2018 Eagle Note was $105,000 as of June 30, 2019. The Company recorded $0 of accrued interest and the total principal outstanding under the November 2018 Eagle Note was $0 as of June 30, 2020 following conversion of $105,000 of principal and $12,832 of accrued interest during the year ended June 30, 2020. The November 2018 Eagle Note may be prepaid with certain penalties by the Company until May 29, 2019. No prepayment was made as of such date.
Upon an event of default, principal and accrued interest will become immediately due and payable under the notes. Additionally, upon an event of default, both notes will accrue interest at a default interest rate of 24% per annum or the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions. In April 2020, Eagle Equities agreed to waive the 24% default interest on this note.
December 24, 2018 Securities Purchase Agreement
Effective December 24, 2018, the Company entered into a securities purchase agreement with Eagle Equities, pursuant to which Eagle Equities purchased a convertible promissory note (the “December 2018 Eagle Note”) from the Company in the aggregate principal amount of $126,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Eagle Equities any time after the six-month anniversary of the December 2018 Eagle Note. The transactions contemplated by the purchase agreement closed on December 24, 2018. Pursuant to the terms of the purchase agreement, Eagle Equities deducted $6,000 from the principal payment due under the December 2018 Eagle Note, at the time of closing, to be applied to its legal expenses. The Company used the net proceeds from the December 2018 Eagle Note to repay an outstanding convertible promissory note before such note became convertible.
| F-18 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
The maturity date of the December 2018 Eagle Note was December 24, 2019. The December 2018 Eagle Note shall bear interest at a rate of 8% per annum, which interest shall be paid by the Company to Eagle Equities in shares of common stock upon receipt of a notice of conversion by the Company from Eagle Equities at any time after the six-month anniversary of the December 2018 Eagle Note.
Additionally, Eagle Equities has the option to convert all or any amount of the principal amount of the December 2018 Eagle Note, at any time, for shares of the Company’s common stock at a price equal to 61% of the lowest closing bid price (the “Closing Bid Price”) of the Company’s common stock as reported on the OTC Markets Group, Inc. quotation system for the ten prior trading days, including the day upon which the Company receives a notice of conversion from Eagle Equities (the “Conversion Price”). However, in the event that the Company’s common stock is restricted by the Depository Trust Company for any reason, the Conversion Price shall be lowered to 51% of the lowest Closing Bid Price for the duration of such restriction. If the Company fails to maintain a reserve of shares of its common stock at least two and a half times the number of shares issuable upon conversion of the December 2018 Eagle Note for at least 60 days after the issuance of the December 2018 Eagle Note, the conversion discount shall be increased by 10%. Notwithstanding the foregoing, Eagle Equities shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Eagle Equities and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock. The December 2018 Eagle Note is treated as stock settled debt under ASC 480 and accordingly, the Company recorded an $80,557 put premium.
The Company recorded $5,220 of accrued interest and the total principal outstanding under the November 2018 Eagle Note was $126,000 as of June 30, 2019. The Company has recorded $15,327 of accrued interest and the total principal outstanding under the November 2018 Eagle Note was $126,000 as of June 30, 2020. The December 2018 Eagle Note may be prepaid with certain penalties until June 22, 2019. No prepayment was made as of such date.
Upon an event of default, principal and accrued interest will become immediately due and payable under the notes. Additionally, upon an event of default, both notes will accrue interest at a default interest rate of 24% per annum or the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions. In April 2020, Eagle Equities agreed to waive the 24% default interest on this note, however, the note is currently past due.
The total principal amount outstanding under the above Eagle Equities financing agreements, specifically the December 12, 2016, the January 27, 2017, the March 1, 2017, the October 25, 2017, the August 9, 2017, the December 29, 2017, the June 14, 2018, the July 13, 2018, the August 29, 2018, the October 2, 2018, the November 30, 2018 and the December 24, 2018 agreements was $792,965 and accrued interest totaled $55,675 as of June 30, 2019. The total principal amount outstanding under the above Eagle Equities financing agreements, specifically the August 29, 2018 and the December 24, 2018 agreements was $205,500 as of June 30, 2020 and accrued interest totaled $26,990.
GS Capital Financing Agreements
July 24, 2017 Securities Purchase Agreement
On July 24, 2017, the Company entered into a Securities Purchase Agreement with GS Capital, pursuant to which GS Capital purchased two 8% convertible redeemable junior subordinated promissory notes, each in the principal amount of $160,000. The first note (the “July 2017 GS Note”) was funded with cash and the second note (the “July 2017 GS Back-End Note”) was initially paid for by an offsetting promissory note issued by GS Capital to the Company (the “July 2017 GS Note Receivable”). The terms of the July 2017 GS Back-End Note required cash funding prior to any conversion thereunder. The July 2017 GS Note Receivable was due March 24, 2018, unless certain conditions were not met, in which case both the July 2017 GS Back-End Note and the July 2017 GS Note Receivable may both be cancelled. Both the July 2017 GS Note and the July 2017 GS Back-End Note matured on July 24, 2018 upon which any outstanding principal and interest is due and payable. The amounts cash funded plus accrued interest under both the July 2017 GS Note and the July 2017 GS Back-End Note are convertible into common stock of the Company at a conversion price equal to 62% of the lowest closing bid price of the common stock for the ten trading days prior to the conversion, subject to adjustment in certain events. On January 25, 2018, the Company received payment of the July 2017 GS Note Receivable in the amount of $160,000 that offset the July 2017 GS Back-End Note. Proceeds from the July 2017 GS Note Receivable of $8,000 were paid directly to legal fees resulting in net cash proceeds of $152,000 received by the Company. As a result, the July 2017 GS Back-End Note is now convertible. The July 2017 GS Note and the July 2017 GS Back-End Note are treated as stock settled debt under ASC 480 and accordingly the Company recorded a $98,065 put premium as each of the notes was funded.
As of June 30, 2018, the outstanding principal under the July 2017 GS Note and $8,169 of accrued interest was fully converted into shares of the Company’s common stock. As of June 30, 2018, $125,000 of principal under the July 2017 GS Back-End Note along with $3,420 in interest was converted. As of June 30, 2019, the remaining outstanding principal balance of $35,000 under the July 2017 GS Back-End Note along with $5,829 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. There was no outstanding balance as of June 30, 2019.
| F-19 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
September 21, 2017 Securities Purchase Agreement
On September 21, 2017, the Company entered into Securities Purchase Agreements, with GS Capital, dated as of September 12, 2017, pursuant to which GS Capital purchased two 8% convertible redeemable junior subordinated promissory notes, each in the principal amount of $160,000. The first note (the “September 2017 GS Note”) was funded with cash and the second note (the “September 2017 GS Back-End Note”) was initially paid for by an offsetting promissory note issued by GS Capital to the Company (the “September 2017 GS Note Receivable”). The terms of the September 2017 GS Back-End Note require cash funding prior to any conversion thereunder. The September 2017 GS Note Receivable was due March 24, 2018, unless certain conditions are not met, in which case both the September 2017 GS Back-End Note and the September 2017 GS Note Receivable may both be cancelled. Both the September 2017 GS Note and the September 2017 GS Back-End Note matured on September 12, 2018, upon which any outstanding principal and interest is due and payable. The amounts cash funded plus accrued interest under both the September 2017 GS Note and the September 2017 GS Back-End Note are convertible into common stock of the Company at a conversion price equal to 62% of the lowest closing bid price of the common stock for the ten trading days prior to the conversion, subject to adjustment in certain events. On February 27, 2018, the Company received payment of the September 2017 GS Note Receivable in the amount of $160,000 that offset the September 2017 GS Back-End Note. Proceeds from the September 2017 GS Note Receivable of $8,000 were paid directly to legal fees resulting in net cash proceeds of $152,000 received by the Company. As a result, the September 2017 GS Back-End Note is now convertible. The September 2017 GS Note and the September 2017 GS Back-End Note are treated as stock settled debt under ASC 480 and accordingly the Company recorded a $98,065 put premium as each of the notes was funded.
As of June 30, 2018, $30,000 of principal under the September 2017 GS Note along with $1,289 in interest was converted. As of June 30, 2019, the remaining outstanding principal balance of $130,000 under the September 2017 GS Note along with $9,695 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. As of June 30, 2019, the outstanding principal balance of $160,000 under the September 2017 GS Back-End note along with $7,119 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. There was no outstanding balance as of June 30, 2019.
March 23, 2018 Securities Purchase Agreement
On March 23, 2018, the Company entered into a securities purchase agreement with GS Capital, pursuant to which GS Capital purchased two 8% convertible redeemable junior subordinated promissory notes of the Company, each in the principal amount of $106,000. The first note (the “March 2018 GS Note”) was funded with cash and the second note (the “March 2018 GS Back-End Note”) was initially paid for by an offsetting promissory note issued by GS Capital to the Company (the “March 2018 GS Note Receivable”). The terms of the March 2018 GS Back-End Note require cash funding prior to any conversion thereunder. The March 2018 GS Note Receivable is due November 23, 2018, unless certain conditions are not met, in which case both the March 2018 GS Back-End Note and the March 2018 GS Note Receivable may both be cancelled. Both the March 2018 GS Note and the March 2018 GS Back-End Note mature on March 23, 2019, upon which any outstanding principal and interest is due and payable. The amounts cash funded plus accrued interest under both the March 2018 GS Note and the March 2018 GS Back-End Note are convertible into shares of common stock of the Company at a conversion price equal to 62% of the lowest closing bid price of the common stock for the ten trading days prior to the conversion, subject to adjustment in certain events. On May 31, 2018, the Company received payment of the March 2018 GS Note Receivable in the amount of $106,000 that offset the March 2018 GS Back-End Note. Proceeds from the March 2018 GS Note Receivable of $5,300 were paid directly to legal fees resulting in net cash proceeds of $100,700 received by the Company. As a result, the March 2018 GS Back-End Note is now convertible. The March 2018 GS Note and the March 2018 GS Back-End Note are treated as stock settled debt under ASC 480 and accordingly the Company recorded a $64,968 put premium as each of the notes was funded.
As of June 30, 2019, the outstanding principal balance of $106,000 under the March 2018 GS Note along with $2,765 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. As of June 30, 2019, the outstanding principal balance of $106,000 under the March 2018 GS Back-End note along with $4,740 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. There was no outstanding balance as of June 30, 2019.
April 13, 2018 Securities Purchase Agreement
On April 13, 2018, the Company entered into a securities purchase agreement with GS Capital, pursuant to which GS Capital purchased two 8% unsecured convertible promissory notes (the “April 2018 GS Notes”) from the Company each in the principal amount of $150,000. The first note (the “April 2018 GS Note”) was funded with cash and the second note (the “April 2018 GS Back-End Note”) was initially paid for by an offsetting promissory note issued by GS Capital to the Company (the “April 2018 GS Note Receivable”). The terms of the April 2018 Back-End Note require cash funding prior to any conversion thereunder.
| F-20 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Both the April 2018 GS Note and the April 2018 GS Back-End Note mature on April 13, 2019, upon which any outstanding principal and interest thereon is due and payable. The amounts cash funded plus accrued interest under both the April 2018 GS Note and the April 2018 GS Back-End Note are convertibles into shares of the Company’s common stock, at any time after October 13, 2018, at a conversion price for each share of common stock equal to 61% of the lowest closing bid price of the Company’s common stock for the ten prior trading days including the day upon which a notice of conversion is received by the Company from GS Capital, subject to adjustment in certain events. On September 12, 2018, the Company received payment of the April 2018 GS Note Receivable in the amount of $150,000 that offset the March 2018 GS Back-End Note. Proceeds from the March 2018 GS Note Receivable of $7,500 were paid directly to legal fees resulting in net cash proceeds of $142,500 received by the Company. Both the April 2018 GS Note and the April 2018 GS Back-End Note are treated as stock settled debt under ASC 480 and accordingly the Company recorded a $95,902 put premium as each of the notes were funded.
As of June 30, 2019, the outstanding principal balance of $150,000 under the April 2018 GS Note along with $9,632 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. As of June 30, 2019, the outstanding principal balance of $150,000 under the April 2018 GS Back-End Note along with $1,606 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. There was no outstanding balance as of June 30, 2019.
October 2, 2018 Securities Purchase Agreement
Effective October 2, 2018, the Company entered into a securities purchase agreement with GS Capital, pursuant to which GS Capital purchased two 8% unsecured convertible redeemable notes (the “October 2, 2018 GS Notes”) from the Company in the aggregate principal amount of $212,000, such principal and the interest thereon convertible into shares of the Company’s common stock. The purchase price of $106,000 of the first note (the “October 2018 GS Note”) was paid in cash by GS Capital on October 3, 2018. After payment of certain legal fees and expenses, net proceeds to the Company from the October 2018 GS Note totaled $100,700. The purchase price of $106,000 of the second note (the “October 2, 2018 GS Back End Note”) was initially paid for by GS Capital issuing to the Company an offsetting $106,000 collateralized secured note (the “October 2, 2018 GS Secured Note”). The terms of the October 2018 GS Back End Note require cash funding prior to any conversion thereunder, and such cash funding shall occur on or before June 2, 2019.
Both the October 2, 2018 GS Note and the October 2, 2018 GS Back End Note, which was funded on February 27, 2019, matured on October 2, 2019, upon which any outstanding principal and interest thereon is due and payable. The amounts cash funded plus accrued interest under both the October 2018 GS Note and the October 2018 GS Back End Note are convertibles into shares of the Company’s common stock, at any time after April 2, 2019, at a conversion price for each share of common stock equal to 61% of the lowest closing bid price of the Company’s common stock for the ten prior trading days including the day upon which a notice of conversion is received by the Company from GS Capital, subject to adjustment in certain events. GS Capital shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by GS Capital and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock. The October 2018 GS Note and the October 2018 GS Back End Note are treated as stock settled debt under ASC 480 and accordingly, the Company recorded a total $67,771 put premium for each note of which $44,690 was released in respect of the October 2018 GS Note in the fiscal year ended June 30, 2019, and a further $22,901 was released during the year ended June 30, 2020 following full conversion of the October 2018 GS Note resulting from conversion of the remaining principal balance of $35,820 and $2,434 in accrued interest. $67,770 of the put premium was released in respect of the October 2018 GS Back-End Note during the year ended June 30, 2020 following conversion $106,000 of the principal balance.
The total principal amount outstanding under the October 2018 GS Note, was $35,820 and accrued interest thereunder totaled $8,531 as of June 30, 2019 and was fully converted during the year ended June 30, 2020 with $3,601 of accrued interest remaining as of June 30, 2020 (see Note 8 – Stockholders’ Deficit).
The maturity date of the October 2, 2018 GS Back-Note was October 2019. The total principal balance under the October 2018 GS Back-End Note, was $106,000 and accrued interest thereunder totaled $5,715 as of June 30, 2019 and the principal balance was $0 and accrued interest totaled $2,658 as of June 30, 2020 (see Note 8 – Stockholders’ Deficit).
The October 2, 2018 GS Notes contain certain events of default, upon which principal and accrued interest will become immediately due and payable. In addition, upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 24% per annum, or if such rate is usurious or not permitted by current law, then at the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
| F-21 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
January 22, 2020 GS Capital Securities Purchase Agreements
Effective January 22, 2020, the Company entered into a securities purchase agreement with GS Capital, pursuant to which GS Capital purchased a convertible promissory note (the “January 22, 2020 GS Note”) from the Company in the aggregate principal amount of $58,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of GS Capital any time after the six month anniversary of the January 22, 2020 GS Capital Note. The January 22, 2020 GS Note contains an original discount of $3,500. The transactions contemplated by the GS Capital Securities Purchase Agreement closed on January 22, 2020. Pursuant to the terms of the GS Capital Securities Purchase Agreement, GS Capital deducted $2,500 from the principal payment due under the January 22, 2020 GW Note, at the time of closing, to be applied to its legal expenses and received net cash proceeds of $52,000 on January 28, 2020. The Company intends to use the net proceeds from the January 22, 2020 GW Note for general working capital purposes.
The maturity date of the January 22, 2020 GS Capital is January 22, 2021. The January 22, 2020 GS Capital Note bears interest at a rate of 10% per annum, which interest may be paid by the Company to GS Capital in shares of the Company’s common stock; but shall not be payable until the January 22, 2020 GS Capital Note becomes payable, whether at the maturity date or upon acceleration or by prepayment.
Additionally, GS Capital has the option to convert all or any amount of the principal face amount of the January 22, 2020 GS Capital Note at any time from the date of issuance and ending on the later of the maturity date or the date the Default Amount is paid if an event of default occurs, which is an amount between 112% and 130% of an amount equal to the then outstanding principal amount of the January 22, 2020 GS Capital Note plus any interest accrued, for shares of the Company’s common stock at the then-applicable conversion price.
The conversion price for the January 22, 2020 GS Capital Note shall be equal to a 40% discount of the lowest closing bid price (“Lowest Trading Price”) of the Common Stock for the ten trading days immediately prior to the delivery of a Notice of Conversion, including the day upon which a Notice of Conversion is received. Notwithstanding the foregoing, GS Capital shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by GS Capital and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock which may be increased up to 9.99% upon 60 days prior written notice by the GS Capital to the Company. The note is treated as stock settled debt under ASC 480 and accordingly the Company recorded a $38,667 put premium which was expensed in fiscal 2020.
The January 22, 2020 GS Note contain certain events of default, upon which principal and accrued interest will become immediately due and payable. In addition, upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 24% per annum, or if such rate is usurious or not permitted by current law, then at the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
The total principal amount outstanding under the above GS Capital financing agreement, specifically the July 24, 2017, the September 21, 2017, the March 23, 2018, the April 13, 2018, and the October 2, 2018 agreements was $141,820 and accrued interest totaled $14,247 as of June 30, 2019. The total principal amount outstanding under the above GS Capital financing agreement, specifically the January 22, 2020 GS Note, was $58,000 and accrued interest of $2,542 as of June 30, 2020.
Convertible Note Issued with Consulting Agreement
August 10, 2017 Consulting Agreement
On August 10, 2017, the Company entered into a consulting agreement, retroactive to May 16, 2017, with a certain consultant, pursuant to which the consultant agreed to provide certain consulting and business advisory services in exchange for a $310,000 junior subordinated convertible note. The maturity date of the August 10, 2017 Convertible Note was August 2019 and is currently past due (see Note 9).The note accrues interest at a rate of 10% per annum and is convertible into common stock at the lesser of $750 or 65% of the three lowest trades in the ten trading days prior to the conversion. The note was fully earned upon signing the agreement and matures on August 10, 2019. The Company accrued $155,000 related to this expense at June 30, 2017 and recorded the remaining $155,000 related to this expense in fiscal year 2018. Upon an event of default, principal and accrued interest will become immediately due and payable under the note. Additionally, upon an event of default, at the election of the holder, the note would accrue interest at a default interest rate of 18% per annum or the highest rate of interest permitted by law. The consulting agreement had a three-month term and expired on August 16, 2017. An aggregate total of $578,212 of this note was bifurcated with the embedded conversion option recorded as a derivative liability at fair value. During the year ended June 30, 2018, the consultant converted $140,000 of principal and $10,764 of interest. During the year ended June 30, 2019, the consultant converted an additional $161,000 of principal and $19,418 of interest leaving a principal balance owed of $9,000 at June 30, 2019. During the year ended June 30, 2020, the consultant converted an additional $500 of principal and $5,248 of interest such that the remaining principal outstanding and accrued interest under this note as of June 30, 2020 was $8,500 and $22,168, respectively.
Power Up Lending Group Financing Agreements
January 22, 2018 Securities Purchase Agreement
Effective January 22, 2018, the Company entered into a securities purchase agreement with Power Up Lending Group Ltd. (“Power Up”), pursuant to which Power Up purchased a convertible promissory note (the “January 2018 Power Up Note”) from the Company in the aggregate principal amount of $153,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Power Up. The transaction closed on January 25, 2018 and the Company received payment on January 29, 2018 in the amount of $153,000, of which $2,500 was paid directly toward legal fees and $500 to Power Up for due diligence fees resulting in net cash proceeds of $150,000.
| F-22 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
The maturity date of the January 2018 Power Up Note was January 22, 2019. The January 2018 Power Up Note bore interest at a rate of 8% per annum, which interest may be paid by the Company to Power Up in shares of the Company’s common stock, but shall not be payable until the January 2018 Power Up Note becomes payable, whether at the maturity date or upon acceleration or by prepayment. An aggregate total of $180,251 of this note was bifurcated with the embedded conversion option recorded as a derivative liability at fair value.
Additionally, Power Up had the option to convert all or any amount of the principal face amount of the January 2018 Power Up Note, starting on July 21, 2018 and ending on the later of the maturity date or the date the Default Amount is paid if an event of default occurs, which was an amount equal to 150% of an amount equal to the then outstanding principal amount of the January 2018 Power Up Note plus any interest accrued for shares of the Company’s common stock at the then-applicable conversion price.
The conversion price for the January 2018 Power Up Note shall be $32.50, subject to certain Market Price (as defined below) adjustment. If the Market Price is greater than or equal to $50.00, the conversion price shall be the greater of 65% of the Market Price (“Variable Conversion Price”) and $32.50. In the event Market Price is less than $50.00, the conversion price shall be the Variable Conversion Price. As defined in the January 2018 Power Up Note, the “Market Price” shall be the average of the lowest three closing bid prices during the ten day trading period prior to and including the day the Company receives a notice of conversion from Power Up on the electronic quotation system or applicable principal securities exchange or trading market or, if no closing bid price of such security is available in any of the foregoing manners, the average of the closing bid prices of any market makers for such security that are listed in the “pink sheets” during the ten prior trading days, including the day upon which the Company receives a notice of conversion from Power Up. Notwithstanding the foregoing, Power Up shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Power Up and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock.
During the fiscal year ended June 30, 2019, the outstanding principal balance of $153,000 along with $6,185 of accrued interest was converted into shares of the Company’s common stock (See Note 8 – Stockholders’ Deficit) resulting in a full repayment of the note.
March 5, 2018 Securities Purchase Agreement
On March 5, 2018, the Company entered into a securities purchase agreement with Power Up, pursuant to which Power Up purchased a convertible promissory note (the “March 2018 Power Up Note”) from the Company in the aggregate principal amount of $53,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Power Up. The Company received payment on March 12, 2018 in the amount of $53,000, of which $2,500 was paid directly toward legal fees and $500 to Power Up for due diligence fees resulting in net cash proceeds of $50,000.
The maturity date of the March 2018 Power Up Note was March 5, 2019. The March 2018 Power Up Note bore interest at a rate of 8% per annum, which interest may be paid by the Company to Power Up in shares of the Company’s common stock, but shall not be payable until the March 2018 Power Up Note becomes payable, whether at the maturity date or upon acceleration or by prepayment. An aggregate total of $65,231 of this note was bifurcated with the embedded conversion option recorded as a derivative liability at fair value.
Additionally, Power Up has the option to convert all or any amount of the principal face amount of the March 2018 Power Up Note, starting on September 1, 2018 and ending on the later of the maturity date or the date the Default Amount is paid if an event of default occurs, which was an amount equal to 150% of an amount equal to the then outstanding principal amount of the March 2018 Power Up Note plus any interest accrued for shares of the Company’s common stock at the then-applicable conversion price.
The conversion price for the March 2018 Power Up Note shall be $32.50, subject to certain Market Price (as defined below) adjustment. If the Market Price is greater than or equal to $50.00, the conversion price shall be the greater of 65% of the Market Price (the “Variable Conversion Price”) and $32.50. In the event Market Price is less than $50.00, the conversion price shall be the Variable Conversion Price. As defined in the March 2018 Power Up Note, the “Market Price” shall be the average of the lowest three closing bid prices during the ten day trading period prior to and including the day the Company receives a notice of conversion from Power Up on the electronic quotation system or applicable principal securities exchange or trading market or, if no closing bid price of such security is available in any of the foregoing manners, the average of the closing bid prices of any market makers for such security that are listed in the “pink sheets” during the ten prior trading days, including the day upon which the Company receives a notice of conversion from Power Up. Notwithstanding the foregoing, Power Up shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Power Up and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock.
The Company had the right to prepay the March 2018 Power Up Note within 180 days of issuance with certain penalties. On August 28, 2018, the Company prepaid the outstanding principal balance of $53,000 and related accrued interest of $2,033 that was due under the March 5, 2018 Power Up Note and the note was deemed fully satisfied. The Company incurred a penalty in the amount of $20,362 as a result of the pre-payment which was reflected in interest expense, there was no outstanding balance as of June 30, 2019.
| F-23 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
May 15, 2018 Securities Purchase Agreement
On May 15, 2018, the Company entered into a securities purchase agreement with Power Up, pursuant to which Power Up purchased a convertible promissory note (the “May 2018 Power Up Note”) from the Company in the aggregate principal amount of $53,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Power Up. The Company received payment on May 18, 2018 in the amount of $53,000, of which $2,500 was paid directly toward legal fees and $500 to Power Up for due diligence fees resulting in net cash proceeds of $50,000.
The maturity date of the May 2018 Power Up Note is May 5, 2019. The May 2018 Power Up Note shall bear interest at a rate of 8% per annum, which interest may be paid by the Company to Power Up in shares of common stock, but shall not be payable until the May 2018 Power Up Note becomes payable, whether at the maturity date or upon acceleration or by prepayment. An aggregate total of $33,744 of this note was bifurcated with the embedded conversion option recorded as a derivative liability at fair value.
Additionally, Power Up had the option to convert all or any amount of the principal face amount of the May 2018 Power Up Note, starting on November 11, 2018 and ending on the later of the maturity date or the date the Default Amount is paid if an event of default occurs, which is an amount equal to 150% of an amount equal to the then outstanding principal amount of the May 2018 Power Up Note plus any interest accrued, for shares of the Company’s common stock at the then-applicable conversion price.
The conversion price for the May 2018 Power Up Note shall be $32.50, subject to certain Market Price (as defined below) adjustment. If the Market Price is greater than or equal to $50.00, the conversion price shall be the greater of 65% of the Market Price (“Variable Conversion Price”) and $32.50. In the event Market Price is less than $50.00, the conversion price shall be the Variable Conversion Price. As defined in the May 2018 Power Up Note, the “Market Price” shall be the average of the lowest three closing bid prices during the ten day trading period prior to and including the day the Company receives a notice of conversion from Power Up on the electronic quotation system or applicable principal securities exchange or trading market or, if no closing bid price of such security is available in any of the foregoing manners, the average of the closing bid prices of any market makers for such security that are listed in the “pink sheets” during the ten prior trading days, including the day upon which the Company receives a notice of conversion from Power Up. Notwithstanding the foregoing, Power Up was restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Power Up and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock.
The Company had the right to prepay the May 2018 Power Up Note within 180 days of issuance with certain penalties. On November 7, 2018, the Company prepaid the outstanding principal balance of $53,000 and related accrued interest of $1,696 that was due under the May 2018 Power Up Note and the note was deemed fully satisfied. The Company incurred a penalty in the amount of $20,715 as a result of the pre-payment which was charged to interest expense. There was no outstanding balance as of June 30, 2019.
August 28, 2018 Securities Purchase Agreement
On August 28, 2018, the Company entered into a securities purchase agreement with Power Up, pursuant to which Power Up purchased a convertible promissory note (the “August 2018 Power Up Note”) from the Company in the aggregate principal amount of $53,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Power Up. The Company received payment on August 29, 2018 in the amount of $53,000, of which $2,500 was paid directly toward legal fees and $500 to Power Up for due diligence fees resulting in net cash proceeds of $50,000.
The maturity date of the August 2018 Power Up Note was August 28, 2019 (the “Maturity Date”). The August 2018 Power Up Note bore interest at a rate of 8% per annum, which interest may be paid by the Company to Power Up in shares of the Company’s common stock, but shall not be payable until the August 2018 Power Up Note becomes payable, whether at the Maturity Date or upon acceleration or by prepayment, as described below.
Additionally, Power Up has the option to convert all or any amount of the principal face amount of the August 2018 Power Up Note, starting on February 24, 2019 at a conversion price of shall be $32.50, subject to certain Market Price (as defined below) adjustment. If the Market Price is greater than or equal to $50.00, the conversion price shall be the greater of 65% of the Market Price (“Variable Conversion Price”) and $32.50. In the event Market Price is less than $50.00, the conversion price shall be the Variable Conversion Price. As defined in the August 2018 Power Up Note, the “Market Price” shall be the average of the lowest three closing bid prices during the ten day trading period prior to and including the day the Company receives a notice of conversion from Power Up on the electronic quotation system or applicable principal securities exchange or trading market or, if no closing bid price of such security is available in any of the foregoing manners, the average of the closing bid prices of any market makers for such security that are listed in the “pink sheets” during the ten prior trading days, including the day upon which the Company receives a notice of conversion from Power Up. Notwithstanding the foregoing, Power Up shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Power Up and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock. An aggregate total of $396,380 of this note was bifurcated with the embedded conversion option recorded as a derivative liability at fair value (See Note 12 - Derivative Financial Instruments and Fair Value Measurements).
| F-24 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 22% per annum. In the event that the Company fails to deliver to Power Up shares of common stock issuable upon conversion of principal or interest under the August 2018 Power Up Note within three business days of a notice of conversion by Power Up, the Company shall incur a penalty of $500, provided, however, that such fee shall not be due if the failure to deliver the shares is a result of a third party such as the transfer agent.
On February 25, 2019, the Company prepaid the outstanding principal balance of $53,000 and related accrued interest of $395 that was due under the August 2018 Power Up Note and the note was deemed fully satisfied. The Company incurred a penalty in the amount of $22,047 as a result of the pre-payment which was charged to interest expense. There was no outstanding balance as of June 30, 2019.
July 3, 2019 Securities Purchase Agreement
Effective July 3, 2019, the Company entered into a securities purchase agreement with Power Up Lending Group Ltd. (“Power Up”), pursuant to which Power Up purchased a convertible promissory note (the “July 3, 2019 Power Up Note”) from the Company in the aggregate principal amount of $78,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Power Up. The transaction closed on July 3, 2019 and the Company received payment on July 8, 2019 in the amount of $78,000, of which $2,500 was paid directly toward legal fees and $500 to Power Up for due diligence fees resulting in net cash proceeds of $75,000.
The maturity date of the July 3, 2019 Power Up Note was July 3, 2020. The July 3, 2019, Power Up Note bore interest at a rate of 8% per annum, which interest may be paid by the Company to Power Up in shares of the Company’s common stock, but shall not be payable until the July 3, 2019 Power Up Note becomes payable, whether at the maturity date or upon acceleration or by prepayment.
Additionally, Power Up had the option to convert all or any amount of the principal face amount of the July 3, 2019 Power Up Note, starting on December 30, 2019 and ending on the later of the maturity date or the date the Default Amount is paid if an event of default occurs, which was an amount equal to 150% of an amount equal to the then outstanding principal amount of the July 3, 2019 Power Up Note plus any interest accrued for shares of the Company’s common stock at the then-applicable conversion price.
The conversion price for the July 3, 2019 Power Up Note shall be $3.25, subject to certain Market Price (as defined below) adjustment. If the Market Price is greater than or equal to $5.00, the conversion price shall be the greater of 65% of the Market Price (“Variable Conversion Price”) and $3.25. In the event Market Price is less than $5.00, the conversion price shall be the Variable Conversion Price. As defined in the July 3, 2019 Power Up Note, the “Market Price” shall be the average of the lowest three closing bid prices during the ten day trading period prior to and including the day the Company receives a notice of conversion from Power Up on the electronic quotation system or applicable principal securities exchange or trading market or, if no closing bid price of such security is available in any of the foregoing manners, the average of the closing bid prices of any market makers for such security that are listed in the “pink sheets” during the ten prior trading days, including the day upon which the Company receives a notice of conversion from Power Up. Notwithstanding the foregoing, Power Up shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Power Up and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock. An aggregate initial total of $155,904 of this note was bifurcated with the embedded conversion option recorded as a derivative liability at fair value (See Note 12 - Derivative Financial Instruments and Fair Value Measurements).
The July 3, 2019 Power Up Note contain certain events of default, upon which principal and accrued interest will become immediately due and payable. In addition, upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 22% per annum, or if such rate is usurious or not permitted by current law, then at the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
The total principal amount outstanding under the above Power Up financing agreement, specifically the July 3, 2019 Power Up Note, was $0 and accrued interest of $0 as of June 30, 2020 following conversion of $78,000 of the principal balance and $3,120 of accrued interest during the year ended June 30, 2020 (see Note 8 - Stockholders’ Deficit).
November 26, 2019 Securities Purchase Agreement
Effective November 26, 2019, the Company entered into a securities purchase agreement with Power Up Lending Group Ltd. (“Power Up”), pursuant to which Power Up purchased a convertible promissory note (the “November 26, 2019 Power Up Note”) from the Company in the aggregate principal amount of $43,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Power Up. The transaction closed on November 22, 2019 and the Company received payment on December 3, 2019 in the amount of $40,000, net of $2,500 paid directly toward legal fees and $500 to Power Up for due diligence fees.
| F-25 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
The maturity date of the November 26, 2019 Power Up Note was November 26, 2020. The November 26, 2019, Power Up Note bore interest at a rate of 8% per annum, which interest may be paid by the Company to Power Up in shares of the Company’s common stock, but shall not be payable until the November 26, 2019 Power Up Note becomes payable, whether at the maturity date or upon acceleration or by prepayment.
Additionally, Power Up has the option to convert all or any amount of the principal face amount of the November 26, 2019 Power Up Note, starting on May 24, 2020 and ending on the later of the maturity date or the date the Default Amount is paid if an event of default occurs, which was an amount equal to 150% of an amount equal to the then outstanding principal amount of the November 26, 2019 Power Up Note plus any interest accrued for shares of the Company’s common stock at the then-applicable conversion price.
The conversion price for the November 26, 2019 Power Up Note shall be $3.05, subject to certain Market Price (as defined below) adjustment. If the Market Price is greater than or equal to $5.00, the conversion price shall be the greater of 65% of the Market Price (“Variable Conversion Price”) and $3.05. In the event Market Price is less than $5.00, the conversion price shall be the Variable Conversion Price. As defined in the November 26, 2019 Power Up Note, the “Market Price” shall be the average of the lowest three closing bid prices during the ten day trading period prior to and including the day the Company receives a notice of conversion from Power Up on the electronic quotation system or applicable principal securities exchange or trading market or, if no closing bid price of such security is available in any of the foregoing manners, the average of the closing bid prices of any market makers for such security that are listed in the “pink sheets” during the ten prior trading days, including the day upon which the Company receives a notice of conversion from Power Up. Notwithstanding the foregoing, Power Up shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Power Up and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock. An aggregate initial total of $52,222 of this note was bifurcated with the embedded conversion option recorded as a derivative liability at fair value (See Note 12 - Derivative Financial Instruments and Fair Value Measurements).
The November 26, 2019 Power Up Note contain certain events of default, upon which principal and accrued interest will become immediately due and payable. In addition, upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 22% per annum, or if such rate is usurious or not permitted by current law, then at the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
The total principal amount outstanding under the above Power Up financing agreement, specifically the November 26, 2019 Power Up Note, was $0 and accrued interest of $0 as of June 30, 2020 following conversion of $43,000 of the principal balance and $1,720 of accrued interest during the year ended June 30, 2020 (see Note 8 - Stockholders’ Deficit).
January 7, 2020 Power Up Lending Group Securities Purchase Agreement
Effective January 7, 2020, the Company entered into a securities purchase agreement with Power Up Lending Group Ltd. (“Power Up”), pursuant to which Power Up purchased a convertible promissory note (the “January 7, 2020 Power Up Note”) from the Company in the aggregate principal amount of $75,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Power Up. The transaction closed on January 7, 2020 and the Company received payment on January 13, 2020 in the amount of $72,000, net of $2,500 paid directly toward legal fees and $500 to Power Up for due diligence fees.
The maturity date of the January 7, 2020 Power Up Note is January 7, 2021. The January 7, 2020, Power Up Note bears interest at a rate of 8% per annum, which interest may be paid by the Company to Power Up in shares of the Company’s common stock, but shall not be payable until the January 7, 2020 Power Up Note becomes payable, whether at the maturity date or upon acceleration or by prepayment.
Additionally, Power Up has the option to convert all or any amount of the principal face amount of the January 7, 2020 Power Up Note, starting on July 4, 2020 and ending on the later of the maturity date or the date the Default Amount is paid if an event of default occurs, which is an amount equal to 150% of an amount equal to the then outstanding principal amount of the January 7, 2020 Power Up Note plus any interest accrued, for shares of the Company’s common stock at the then-applicable conversion price.
The conversion price for the January 7, 2020 Power Up Note shall be $3.05, subject to certain Market Price (as defined below) adjustment. If the Market Price is greater than or equal to $5.00, the conversion price shall be the greater of 65% of the Market Price (“Variable Conversion Price”) and $3.05. In the event Market Price is less than $5.00, the conversion price shall be the Variable Conversion Price. As defined in the January 7, 2020 Power Up Note, the “Market Price” shall be the average of the lowest three closing bid prices during the ten day trading period prior to and including the day the Company receives a notice of conversion from Power Up on the electronic quotation system or applicable principal securities exchange or trading market or, if no closing bid price of such security is available in any of the foregoing manners, the average of the closing bid prices of any market makers for such security that are listed in the “pink sheets” during the ten prior trading days, including the day upon which the Company receives a notice of conversion from Power Up. Notwithstanding the foregoing, Power Up shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Power Up and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock. An aggregate total of $314,406 of this note was bifurcated with the embedded conversion option recorded as a derivative liability at fair value (See Note 12 - Derivative Financial Instruments and Fair Value Measurements).
| F-26 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
The January 7, 2020 Power Up Note contain certain events of default, upon which principal and accrued interest will become immediately due and payable. In addition, upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 22% per annum, or if such rate is usurious or not permitted by current law, then at the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
The total principal amount outstanding under the above Power Up financing agreement, specifically the January 7, 2020 Power Up Note, was $75,000 and accrued interest of $2,869 as of June 30, 2020.
March 12, 2020 Power Up Lending Group Securities Purchase Agreement
Effective March 12, 2020, the Company entered into a securities purchase agreement with Power Up Lending Group Ltd. (“Power Up”), pursuant to which Power Up purchased a convertible promissory note (the “March 12, 2020 Power Up Note”) from the Company in the aggregate principal amount of $43,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Power Up. The transaction closed on March 12, 2020 and the Company received payment on March 5, 2020 in the amount of $40,000, net of $2,500 paid directly toward legal fees and $500 to Power Up for due diligence fees.
The maturity date of the March 12, 2020 Power Up Note is March 12, 2021. The March 12, 2020, Power Up Note bears interest at a rate of 8% per annum, which interest may be paid by the Company to Power Up in shares of the Company’s common stock, but shall not be payable until the March 12, 2020 Power Up Note becomes payable, whether at the maturity date or upon acceleration or by prepayment.
Additionally, Power Up has the option to convert all or any amount of the principal face amount of the March 12, 2020 Power Up Note, starting on September 4, 2020 and ending on the later of the maturity date or the date the Default Amount is paid if an event of default occurs, which is an amount equal to 150% of an amount equal to the then outstanding principal amount of the March 12, 2020 Power Up Note plus any interest accrued, for shares of the Company’s common stock at the then-applicable conversion price.
The conversion price for the March 12, 2020 Power Up Note shall be $3.05, subject to certain Market Price (as defined below) adjustment. If the Market Price is greater than or equal to $5.00, the conversion price shall be the greater of 65% of the Market Price (“Variable Conversion Price”) and $3.05. In the event Market Price is less than $5.00, the conversion price shall be the Variable Conversion Price. As defined in the March 12, 2020 Power Up Note, the “Market Price” shall be the average of the lowest three closing bid prices during the ten day trading period prior to and including the day the Company receives a notice of conversion from Power Up on the electronic quotation system or applicable principal securities exchange or trading market or, if no closing bid price of such security is available in any of the foregoing manners, the average of the closing bid prices of any market makers for such security that are listed in the “pink sheets” during the ten prior trading days, including the day upon which the Company receives a notice of conversion from Power Up. Notwithstanding the foregoing, Power Up shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Power Up and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock. An aggregate initial total of $55,929 of this note was bifurcated with the embedded conversion option recorded as a derivative liability at fair value (See Note 12 - Derivative Financial Instruments and Fair Value Measurements).
The March 12, 2020 Power Up Note contain certain events of default, upon which principal and accrued interest will become immediately due and payable. In addition, upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 22% per annum, or if such rate is usurious or not permitted by current law, then at the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
The total principal amount outstanding under the above Power Up financing agreement, specifically the March 12, 2020 Power Up Note, was $43,000 and accrued interest of $1,034 as of June 30, 2020 (see Note 8 - Stockholders’ Deficit).
JSJ Investments, Inc. Financing Agreement
June 26, 2018 Securities Purchase Agreement
Effective June 26, 2018, the Company issued a convertible promissory note (the “June 2018 JSJ Note”) to JSJ Investments, Inc. (“JSJ”) in the aggregate principal amount of $113,000, with principal and the interest thereon convertible into shares of the Company’s common stock at the option of JSJ any time after 180 days of issuance. At the time of closing on June 27, 2018, JSJ deducted $3,000 from the principal payment due under the June 2018 JSJ Note to be applied to its legal expenses, such that the Company received aggregate net proceeds of $110,000 at closing.
| F-27 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
The maturity date of the June 2018 JSJ Note was June 26, 2019, unless extended for up to one year at JSJ’s discretion (the “Maturity Date”). The June 2018 JSJ Note bore interest at a rate of 8% per annum, and after the maturity date shall compound quarterly.
Additionally, JSJ had the option to convert all or any amount of the principal face amount of the June 2018 JSJ Note, at any time beginning December 23, 2018, for shares of the Company’s common stock at the conversion prices set forth in the note. The June 2018 JSJ Note was treated as stock settled debt under ASC 480 and accordingly the Company recorded a $60,846 put premium.
The Company had the right to prepay the June 2018 JSJ Note until December 23, 2018. If the June 2018 JSJ Note was prepaid within 90 days of the issuance date, then the prepayment premium shall be 135% of the face amount plus any accrued interest; if the JSJ Note was prepaid after 90 days from the issuance date, but prior to 121 days from the issuance date, then the prepayment premium shall be 140% of the face amount plus any accrued interest; and if the June 2018 JSJ Note was prepaid after 120 days from the issuance date, but prior to 180 days from the issuance date, then the prepayment premium shall be 145% of the face amount plus any accrued interest.
On December 24, 2018, the Company prepaid the outstanding principal balance of $113,000 and related accrued interest of $4,508 that was due under the June 2018 JSJ Note resulting in full repayment of the note and a full reduction of the put premium. The Company incurred a penalty in the amount of $51,380 as a result of the pre-payment. There was no outstanding balance as of June 30, 2019.
Coventry Enterprises, LLC Financing Agreement
June 29, 2018 Securities Purchase Agreement
Effective June 29, 2018, the Company entered into a securities purchase agreement with Coventry Enterprises, LLC (“Coventry Enterprises”), pursuant to which Coventry Enterprises purchased two 8% unsecured convertible promissory notes from the Company in the aggregate principal amount of $200,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Coventry Enterprises.
The purchase price of $100,000 of the first note (the “July 2018 Coventry Note”) was paid in cash by Coventry Enterprises on July 2, 2018. After payment of certain legal fees and expenses, net proceeds to the Company from the First Note totaled $95,000. The purchase price of $100,000 of the second note (the “July 2018 Coventry Back-End Note”) was initially paid for by the issuance of an offsetting $100,000 collateralized secured note issued to Company by Coventry Enterprises (the “July 2018 Coventry Enterprises Note”). The terms of the July 2018 Coventry Back-End Note require cash funding prior to any conversion thereunder. The July 2018 Coventry Back-End Note was due February 29, 2019, unless certain conditions were not met, in which case both the July 2018 Coventry Back-End Note and the July 2018 Coventry Enterprise Note may both be cancelled. On September 6, 2018, the Company received payment of the July 2018 Coventry Enterprise Note in the amount of $100,000 that offset the July 2018 Coventry Back-End Note. Proceeds from the July 2018 Coventry Enterprise Note of $5,000 were paid directly to legal fees resulting in net cash proceeds of $95,000 received by the Company. As a result, the July 2018 Coventry Back-End Note then became convertible.
The maturity date of the July 2018 Coventry Note and the July 2018 Coventry Back-End Note was June 29, 2019. The outstanding principal amounts plus accrued interest under both the July 2018 Coventry Note and the July 2018 Coventry Back-End Note were convertible into shares of common stock of the Company at a conversion price equal to 61% of the lowest closing bid price of the Company’s common stock as reported on the exchange or quotation system on which the Company’s shares are then traded for the ten prior trading days including the day upon which a notice of conversion is received by the Company from Coventry Enterprises. Coventry Enterprises shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Coventry Enterprises and its affiliates, exceeds 9.9% of the outstanding shares of the Company’s common stock. Both the July 2018 Coventry Note and the July 2018 Coventry Back-End Notes were treated as stock settled debt under ASC 480 and accordingly the Company recorded a $63,934 put premium as each of the notes was funded.
As of June 30, 2019, the outstanding principal balance of $100,000 under the June 2018 Coventry Enterprises Note along with $7,479 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. As of June 30, 2019, the outstanding principal balance of $100,000 under the June 2018 Coventry Enterprises Back-End Note along with $8,137 of accrued interest was fully converted (see Note 8 – Stockholders’ Deficit) resulting in full repayment of the note and a full reduction of the put premium. There was no outstanding balance as of June 30, 2019.
Upon an event of default, principal and accrued interest will become immediately due and payable under the notes. Additionally, upon an event of default, both notes will accrue interest at a default interest rate of 24% per annum or the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
| F-28 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Redstart Holdings Corp Financing Agreement
May 23, 2019 Securities Purchase Agreement
Effective May 23, 2019, the Company issued a convertible promissory note (the “May 23 Redstart Holdings Note”) to Redstart Holdings Corp (“Redstart Holdings”) in the aggregate principal amount of $133,000, with principal and the interest thereon convertible into shares of the Company’s common stock at the option of Redstart Holdings any time after 180 days of issuance. At the time of closing on May 31, 2019, Redstart Holdings deducted $3,000 from the principal payment due under the May 2019 Redstart Holdings Note to be applied to its legal expenses, such that the Company received aggregate net proceeds of $130,000 at closing.
The maturity date of the May 2019 Redstart Holdings Note was May 23, 2020 and bore interest at a rate of 8% per annum.
Additionally, Redstart Holdings had the option to convert all or any amount of the principal face amount of the May 2019 Redstart Note, starting on November 19, 2019 at a conversion price subject to certain Market Price (as defined below) adjustment. If the Market Price is greater than or equal to $50.00, the conversion price shall be the greater of 65% of the Market Price (“Variable Conversion Price”) and $32.50. In the event Market Price is less than $50.00, the conversion price shall be the Variable Conversion Price. As defined in the May 2019 Redstart Holdings Note, the “Market Price” shall be the average of the lowest three closing bid prices during the ten day trading period prior to and including the day the Company receives a notice of conversion from Redstart Holdings on the electronic quotation system or applicable principal securities exchange or trading market or, if no closing bid price of such security is available in any of the foregoing manners, the average of the closing bid prices of any market makers for such security that are listed in the “pink sheets” during the ten prior trading days, including the day upon which the Company receives a notice of conversion from Redstart Holdings. Notwithstanding the foregoing, Redstart Holdings shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Redstart Holdings and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock. An aggregate total of $166,564 of this note was bifurcated with the embedded conversion option recorded as a derivative liability at fair value (See Note 12 - Derivative Financial Instruments and Fair Value Measurements).
The Company had the right to prepay the May 2019 Redstart Holdings Note until November 19, 2019. If the May 2019 Redstart Holdings Note was prepaid within 90 days of the issuance date, then the prepayment premium shall be 115% of the face amount plus any accrued interest; if the May 2019 Redstart Holdings Note was prepaid after 91 days from the issuance date, but prior to 121 days from the issuance date, then the prepayment premium shall be 120% of the face amount plus any accrued interest; and if the May 2019 Redstart Holdings Note was prepaid after 121 days from the issuance date, but prior to 150 days from the issuance date, then the prepayment premium shall be 125% of the face amount plus any accrued interest; and if the May 2019 Redstart Holdings Note was prepaid after 151 days from the issuance date, but prior to 180 days from the issuance date, then the prepayment premium shall be 129% of the face amount plus any accrued interest.
The May 23, 2019 Redstart Holdings Note contained certain events of default, upon which principal and accrued interest will become immediately due and payable. In addition, upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 22% per annum, or if such rate is usurious or not permitted by current law, then at the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
The total principal amount outstanding and accrued interest under the above Redstart Holdings financing agreement, specifically the May 23, 2019 agreement at June 30, 2019 was $133,000 and $1,137 respectively and as of June 30, 2020 total principal amount outstanding and accrued interest totaled $0 and $0 respectively following conversion of $133,000 of the principal balance and $5,320 of accrued interest during the year ended June 30, 2020.
Odyssey Capital Financing Agreements
July 30, 2019 Securities Purchase Agreement
Effective July 30, 2019, the Company entered into a securities purchase agreement with Odyssey Capital Funding LLC, (“Odyssey”), pursuant to which Odyssey purchased a convertible promissory note (the “July 30, 2019 Odyssey Note”) from the Company in the aggregate principal amount of $320,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Odyssey. The July 30, 2019 Odyssey Note contains an original discount of $25,000. The transaction closed on July 30, 2019 and the Company received payment on August 1, 2019 in the amount of $295,000, of which $10,000 was paid directly toward legal fees, resulting in net cash proceeds of $285,000.
The maturity date of the July 30, 2019 Odyssey Note was July 30, 2020. The July 2019 Odyssey Note bore interest at a rate of 10% per annum, which interest may be paid by the Company to Odyssey in shares of the Company’s common stock, but shall not be payable until the July 30, 2019 Odyssey Note becomes payable, whether at the maturity date or upon acceleration or by prepayment. The note was treated as stock settled debt under ASC 480 and accordingly the Company recorded a $172,308 put premium.
| F-29 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Additionally, Odyssey had the option to convert all or any amount of the principal face amount of the July 30, 2019 Odyssey Note, starting on January 31, 2020 and ending on the later of the maturity date or the date the Default Amount is paid if an event of default occurs, which was an amount equal to 120% of an amount equal to the then outstanding principal amount of the July 30, 2019 Odyssey Note plus any interest accrued from July 30, 2019 at the default interest rate of 24% per annum for shares of the Company’s common stock at the then-applicable conversion price.
The conversion price for the July 30, 2019 Odyssey Note shall be equal to 65% of the lowest closing bid price of the Common Stock as reported on the OTC Markets on which the Company’s shares were then traded or any exchange upon which the Common Stock may be traded in the future, for the ten prior trading days including the day upon which a Notice of Conversion is received by the Company.
Common Stock beneficially owned by the Holder and its affiliates would exceed 4.99% of the outstanding shares of the Common Stock of the Company (which may be increased up to 9.9% upon 60 days’ prior written notice by the Holder to the Company).
The July 30, 2019 Odyssey Note contained certain events of default, upon which principal and accrued interest will become immediately due and payable. In addition, upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 24% per annum, or if such rate is usurious or not permitted by current law, then at the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
The total principal amount outstanding under the above Odyssey financing agreement, specifically the July 30, 2019 Odyssey Note, was $0 and accrued interest of $0 as of June 30, 2020 following conversion of $320,000 of the principal balance and $23,220 of accrued interest during the year ended June 30, 2020 resulting in full repayment of the note and a full reduction of the put premium.
Auctus Fund Financing Agreements
August 30, 2019 Securities Purchase Agreement
Effective August 30, 2019, the Company entered into a securities purchase agreement with Auctus Fund, LLC (“Auctus”), pursuant to which Auctus purchased a convertible promissory note (the “August 30, 2019 Auctus Note”) from the Company in the aggregate principal amount of $550,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Auctus. The transaction closed on August 30, 2019 and the Company received payment on September 4, 2019 in the amount of $550,000, of which $5,000 was paid directly toward legal fees and $40,000 to Auctus for due diligence fees resulting in net cash proceeds of $505,000.
The maturity date of the August 30, 2019 Auctus Note was August 30, 2020 and is past due as of the date of this filing. The August 30, 2019 Auctus Note bears interest at a rate of 10% per annum, but shall not be payable until the August 30, 2019 Auctus Note becomes payable, whether at the maturity date or upon acceleration or by prepayment. The note is treated as stock settled debt under ASC 480 and accordingly the Company recorded a $366,667 put premium. The August 30, 2019 Auctus Note may not be prepaid without the written consent of Auctus. Any amount of principal or interest which is not paid when due shall bear interest at the rate of 24% per annum.
Additionally, Auctus has the option to convert all or any amount of the principal face amount and accrued interest of the August 30, 2019 Auctus Note, at any time following the issue date and ending on the later of the maturity date or the date of payment of the Default Amount if an event of default occurs, which is an amount equal to 125% of an amount equal to the then outstanding principal amount of the August 30, 2019 Auctus Note (but not less than $15,000) plus any interest accrued from August 30, 2019 at the default interest rate of 24% per annum, for shares of the Company’s common stock at the then-applicable conversion price. Upon the holder’s election to convert accrued interest, default interest or any penalty amounts as stipulated, the Company may elect to pay those amounts in cash. The note may also be prepaid by the Company at any time between the date of issuance and August 13, 2020 at 135% multiplied by the sum of (a) the then outstanding principal amount plus (b) accrued and unpaid interest plus (c) default interests, if any.
The conversion price for the August 30, 2019 Auctus Note shall be the Variable Conversion Price, being 60% of the Market Price on the date of conversion. Notwithstanding the foregoing, Auctus shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Auctus and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock.
In connection with the issuance of the August 2019 Auctus Note, the Company issued common stock purchase warrants to Auctus to purchase 450,000 shares of the Company’s common stock (the “First Warrant”) as a commitment fee upon the terms and subject to the limitations and conditions set forth in such First Warrant at an “Exercise Price” of $2.25. In connection with the issuance of the Note, the Company shall issue a common stock purchase warrant to Buyer to purchase 300,000 shares of the Company’s common stock (the “Second Warrant”) as a commitment fee upon the terms and subject to the limitations and conditions set forth in such Second Warrant at an “Exercise Price” of $3.33. In connection with the issuance of the Note, the Company shall issue a common stock purchase warrant to Buyer to purchase 225,000 shares of the Company’s common stock (the “Third Warrant”) as a commitment fee upon the terms and subject to the limitations and conditions set forth in such Third Warrant at an “Exercise Price” of $4.50. The First Warrant, Second Warrant, and Third Warrant shall collectively be referred as the “Warrants”. The Warrants have an “Exercise Period” of five years from the date of issuance being August 30, 2019. Under the terms of the Purchase Agreement and the Warrants, the Selling Security Holder may not either convert the Notes nor exercise the Warrants to the extent (but only to the extent) that the Selling Security Holder or any of its affiliates would beneficially own a number of shares of our Common Stock which would exceed 4.99% of our outstanding shares. The Company accounted for the warrants by using the relative fair value method and recorded debt discount from the relative fair value of the warrants of $375,905 using a simple binomial lattice model (see Note 8).
| F-30 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
In connection with the Purchase Agreement, the Company and the Purchaser entered into a Registration Rights Agreement (the “Registration Rights Agreement”). Pursuant to the Registration Rights Agreement, the Company agreed to register the shares of Common Stock underlying the Securities in a Registration Statement with the SEC as well as the Commitment Shares (as defined herein). The Registration Rights Agreement contains customary representations, warranties, agreements and indemnification rights and obligations of the parties.
The Note is subject to customary default provisions and also includes a cross-default provision which provides that a breach or default by the Borrower of any covenant or other term or condition contained in any of the Other Agreements (as defined therein), after the passage of all applicable notice and cure or grace periods, shall, at the option of the Holder, be considered a default under this Note and the Other Agreements. Upon occurrence of any such event, the Holder shall be entitled (but in no event required) to apply all rights and remedies of the Holder under the terms of this Note and the Other Agreements by reason of a default under said Other Agreements or the Note.
The August 30, 2019 Auctus Note contain certain events of default, upon which principal and accrued interest will become immediately due and payable. In addition, upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 24% per annum, or if such rate is usurious or not permitted by current law, then at the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
The total principal amount outstanding under the above Auctus financing agreement, specifically the August 30, 2019 Auctus Note, was $358,965 and accrued interest of $486 as of June 30, 2020 following conversion of $191,035 of the principal balance and $43,176 of accrued interest during the year ended June 30, 2020. Accordingly, $127,356 of the put premium was released in respect of the August 30, 2019 Auctus Note during the year ended June 30, 2020 following conversion of the principal balance.
GW Holdings Securities Purchase Agreements
Effective October 1, 2019, the Company entered into a securities purchase agreement with GW Holdings, pursuant to which GW Holdings purchased a convertible promissory note (the “October 1, 2019 GW Note”) from the Company in the aggregate principal amount of $131,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of GW Holdings any time after the six month anniversary of the October 1, 2019 GW Holdings Note. The transactions contemplated by the GW Holdings Securities Purchase Agreement closed on October 1, 2019. Pursuant to the terms of the GW Holdings Securities Purchase Agreement, Eagle Equities deducted $6,000 from the principal payment due under the October 1, 2019 GW Note, at the time of closing, to be applied to its legal expenses. The Company intends to use the net proceeds of $125,000 from the October 1, 2019 GW Note for general working capital purposes.
The maturity date of the October 1, 2019 GW Holdings is October 1, 2020. The October 1, 2019 GW Holdings Note bears interest at a rate of 8% per annum, which interest may be paid by the Company to GW Holdings in shares of the Company’s common stock; but shall not be payable until the October 1, 2019 GW Holdings Note becomes payable, whether at the maturity date or upon acceleration or by prepayment.
Additionally, GW Holdings has the option to convert all or any amount of the principal face amount of the October 1, 2019 GW Holdings Note at any time from the date of issuance and ending on the later of the maturity date or the date the Default Amount is paid if an event of default occurs, which is an amount between 110% and 150% of an amount equal to the then outstanding principal amount of the October 1, 2019 GW Holdings Note plus any interest accrued, for shares of the Company’s common stock at the then-applicable conversion price.
The conversion price for the October 1, 2019 GW Holdings Note shall be equal to a 40% discount of the lowest closing bid price (“Lowest Trading Price”) of the Common Stock for the ten trading days immediately prior to the delivery of a Notice of Conversion, including the day upon which a Notice of Conversion is received. Notwithstanding the foregoing, GW Holdings shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by GW Holdings and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock which may be increased up to 9.99% upon 60 days prior written notice by the GW Holdings to the Company. The note is treated as stock settled debt under ASC 480 and accordingly the Company recorded a $87,333 put premium.
The October 1, 2019 GW Holdings Note contain certain events of default, upon which principal and accrued interest will become immediately due and payable. In addition, upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 24% per annum, or if such rate is usurious or not permitted by current law, then at the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
The total principal amount outstanding under the above GW Holdings financing agreement, specifically the October 1, 2019 GW Holdings Note, was $30,000 and accrued interest of $1,776 as of June 30, 2020 following conversion of $101,000 of the principal balance and $5,082 of accrued interest during the year ended June 30, 2020. Accordingly, $67,333 of the put premium was reclassed to additional paid in capital in respect of the October 1, 2019 GW Holdings Note during the year ended June 30, 2020 following conversion of the principal balance.
| F-31 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Crown Bridge Securities Purchase Agreements
Effective October 3, 2019, the Company entered into a securities purchase agreement with Crown Bridge Partners, pursuant to which Crown Bridge purchased a convertible promissory note (the “October 3, 2019 Crown Bridge Note”) from the Company in the aggregate principal amount of $108,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Crown Bridge any time from the of issuance of the of the October 3, 2019 Crown Bridge Note. The transactions contemplated by the Crown Bridge Securities Purchase Agreement closed on October 3, 2019. Pursuant to the terms of the Crown Bridge Securities Purchase Agreement, Crown Bridge deducted $3,000 from the principal payment due under the October 3, 2019 Crown Bridge Note, at the time of closing, to be applied to its legal expenses, and there was a $5,000 original issuance discount resulting in $100,000 net proceeds to the Company. The Company intends to use the net proceeds from the October 3, 2019 Crown Bridge Note for general working capital purposes.
The maturity date of the October 3, 2019 Crown Bridge is October 3, 2020. The October 3, 2019 Crown Bridge Note bears interest at a rate of 10% per annum, which interest may be paid by the Company to Crown Bridge in shares of the Company’s common stock; but shall not be payable until the October 2019 Crown Bridge Note becomes payable, whether at the maturity date or upon acceleration or by prepayment.
Additionally, Crown Bridge has the option to convert all or any amount of the principal face amount of the October 3, 2019 Crown Bridge Note at any time from the date of issuance and ending on the later of the maturity date or the date the Default Amount is paid if an event of default occurs, which is an amount between 110% and 150% of an amount equal to the then outstanding principal amount of the October 3, 2019 Crown Bridge Note plus any interest accrued, for shares of the Company’s common stock at the then-applicable conversion price.
The conversion price for the October 3, 2019 Crown Bridge Note shall be equal to a 40% discount of the lowest closing bid price (“Lowest Trading Price”) of the Common Stock for the ten trading days immediately prior to the delivery of a Notice of Conversion, including the day upon which a Notice of Conversion is received. Notwithstanding the foregoing, Crown Bridge shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Crown Bridge and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock which may be increased up to 9.99% upon 60 days prior written notice by the Crown Bridge to the Company. The note is treated as stock settled debt under ASC 480 and accordingly the Company recorded a $72,000 put premium.
The October 3, 2019 Crown Bridge Note contain certain events of default, upon which principal and accrued interest will become immediately due and payable. In addition, upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 15% per annum, or if such rate is usurious or not permitted by current law, then at the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
The total principal amount outstanding under the above Crown Bridge financing agreement, specifically the October 3, 2019 Crown Bridge Note, was $65,280 and accrued interest of $7,232 as of as of June 30, 2020 following conversion of $42,720 of the principal balance during the year ended June 30, 2020. Accordingly, $28,480 of the put premium was released in respect of the October 3, 2019 Crown Bridge Note during the year ended June 30, 2020 following conversion of the principal balance.
Ader Alef Securities Purchase Agreements
Effective January 13, 2020, the Company entered into a securities purchase agreement with Ader Alef, pursuant to which Ader Alef purchased a convertible promissory note (the “January 13, 2020 Ader Alef Note”) from the Company in the aggregate principal amount of $110,250, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of Ader Alef any time after the six month anniversary of the January 13, 2020 Ader Alef Note. The January 13, 2020 Ader Alef Note contains an original discount of $5,250. The transactions contemplated by the Ader Alef Securities Purchase Agreement closed on January 13, 2020. Pursuant to the terms of the Ader Alef Securities Purchase Agreement, Ader Alef deducted $5,000 from the principal payment due under the January 13, 2020 Ader Alef Note at the time of closing, to be applied to its legal expenses and the Company received net cash proceeds of $100,000 on January 15, 2020. The Company intends to use the net proceeds from the January 13, 2020 Ader Alef Note for general working capital purposes.
The maturity date of the January 13, 2020 Ader Alef is January 13, 2021. The January 13, 2020 Ader Alef Note bears interest at a rate of 8% per annum, which interest may be paid by the Company to Ader Alef in shares of the Company’s common stock; but shall not be payable until the January 13, 2020 Ader Alef Note becomes payable, whether at the maturity date or upon acceleration or by prepayment.
Additionally, Ader Alef has the option to convert all or any amount of the principal face amount of the January 13, 2020 Ader Alef Note at any time from the date of issuance and ending on the later of the maturity date or the date the Default Amount is paid if an event of default occurs, which is an amount between 120% and 150% of an amount equal to the then outstanding principal amount of the January 13, 2020 Ader Alef Note plus any interest accrued, for shares of the Company’s common stock at the then-applicable conversion price.
| F-32 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
The conversion price for the January 13, 2020 Ader Alef Note during the first 6 months the January 13, 2020 Ader Alef Note is in effect shall be fixed at $2.50 and thereafter shall be equal to a 35% discount of the lowest closing bid price (“Lowest Trading Price”) of the Common Stock for the ten trading days immediately prior to the delivery of a Notice of Conversion, including the day upon which a Notice of Conversion is received. Notwithstanding the foregoing, Ader Alef shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by Ader Alef and its affiliates, exceeds 4.99% of the outstanding shares of the Company’s common stock which may be increased up to 9.99% upon 60 days prior written notice by the Ader Alef to the Company. The note is treated as stock settled debt under ASC 480 and accordingly the Company recorded a $59,365 put premium.
The January 13, 2020 Ader Alef Note contain certain events of default, upon which principal and accrued interest will become immediately due and payable. In addition, upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 24% per annum, or if such rate is usurious or not permitted by current law, then at the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
The total principal amount outstanding under the above Ader Alef financing agreement, specifically the January 13, 2020 Ader Alef Note, was $110,250 and accrued interest of $4,073 as of June 30, 2020.
LG Capital Securities Purchase Agreements
Effective February 19, 2020, the Company entered into a securities purchase agreement with LG Capital Funding, LLC (“LG Capital”), pursuant to which LG Capital purchased a convertible promissory note (the “February 19, 2020 LG Capital Note”) from the Company in the aggregate principal amount of $75,000, such principal and the interest thereon convertible into shares of the Company’s common stock at the option of LG Capital any time after the six month anniversary of the February 19, 2020 LG Capital Note. The February 19, 2020 LG Capital Note contains an original discount of $3,750. The transactions contemplated by the LG Capital Securities Purchase Agreement closed on March 4, 2020. Pursuant to the terms of the LG Capital Securities Purchase Agreement, LG Capital deducted $2,500 from the principal payment due under the February 19, 2020 LG Capital Note at the time of closing, to be applied to its legal expenses and the Company received net cash proceeds of $71,250 on March 25, 2020. The Company intends to use the net proceeds from the February 19, 2020 LG Capital Note for general working capital purposes.
The maturity date of the February 19, 2020 LG Capital Note is February 19, 2021. The February 19, 2020 LG Capital Note bears interest at a rate of 8% per annum, which interest may be paid by the Company to LG Capital in shares of the Company’s common stock; but shall not be payable until the February 19, 2020 LG Capital Note becomes payable, whether at the maturity date or upon acceleration or by prepayment.
During the first 60 to 180 days following the date of the note, the Company has the right to prepay the principal and accrued but unpaid interest due under the February 19, 2020 LG Capital Note, together with any other amounts that the Company may owe the holder under the terms of the note, at a premium ranging from 112% to 135% as defined in the note agreement. After this initial 180-day period, the Company does not have a right to prepay the February 19, 2020 LG Capital Note.
The conversion price for the February 19, 2020 LG Capital Note during the first 6 months the February 19, 2020 LG Capital Note is in effect shall be fixed at $0.50 and thereafter shall be equal to a 35% discount of the lowest closing bid price (“Lowest Trading Price”) of the Common Stock for the ten trading days immediately prior to the delivery of a Notice of Conversion, including the day upon which a Notice of Conversion is received. Notwithstanding the foregoing, LG Capital shall be restricted from effecting a conversion if such conversion, along with other shares of the Company’s common stock beneficially owned by LG Capital and its affiliates, exceeds 9.99% of the outstanding shares of the Company’s common stock. The note is treated as stock settled debt under ASC 480 and accordingly the Company recorded a $40,385 put premium.
The February 19, 2020 LG Capital Note contain certain events of default, upon which principal and accrued interest will become immediately due and payable. In addition, upon an event of default, interest on the outstanding principal shall accrue at a default interest rate of 24% per annum, or if such rate is usurious or not permitted by current law, then at the highest rate of interest permitted by law. Further, certain events of default may trigger penalty and liquidated damage provisions.
The total principal amount outstanding under the above LG Capital financing agreement, specifically the February 19, 2020 LG Capital Note, was $75,000 and accrued interest of $2,164 as of June 30, 2020.
Amortization of debt discounts
The Company recorded $728,904 and $243,850 of debt discounts (including warrants, derivatives, debt issue costs and original issue discounts) related to the above note issuances during the years ended June 30, 2020 and 2019, respectively. The Company recorded $836,724 and $767,000 of put premiums related to the above note issuances during the years ended June 30, 2020 and 2019, respectively. The debt discounts are being amortized over the term of the debt and the put premiums are expensed on issuance of the debt with the liability released to additional paid in capital on conversion of the principal.
| F-33 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Amortization of all debt discounts for the years ended June 30, 2020 and 2019 was $734,130 and $389,673, respectively.
The Company reclassified $874,924 and $1,824,317 in put premiums to additional paid in capital following conversions during the years ended June 30, 2020 and 2019, respectively.
NOTE 7 – INCOME TAXES
Through June 30, 2010, the Company operated exclusively in Australia. The Company was wholly subject to Australian income tax laws and regulations, which are administered by the Australian Taxation Office for the years ended June 30, 2010 and all prior years.
On November 23, 2010, the Company was incorporated in the state of Delaware. In January 2011, the Company acquired all of the outstanding shares of Propanc PTY LTD on a one-for-one basis with Propanc PTY LTD becoming a wholly owned subsidiary of the Company. As a result of these transactions, the Company is subject to the income tax laws of both the United States and Australia for the years ended June 30, 2013 through June 30, 2020.
The reconciliation of income tax expense computed at the U.S. federal statutory rate of 21% to the income tax provision for the years ended June 30, 2020 and 2019 is as follows:
| Year Ended | ||||||||
| June 30, 2020 | June 30, 2019 | |||||||
| Taxes under statutory US tax rates | $ | (995,552 | ) | $ | (1,209,258 | ) | ||
| Increase (decrease) in valuation allowance | 1,137,716 | 1,394,444 | ||||||
| Prior period adjustment | (14,624 | ) | - | |||||
| Foreign tax rate differential | (128,492 | ) | (186,286 | ) | ||||
| Other | 952 | 1,100 | ||||||
| Income tax (expense) benefit | $ | - | $ | - | ||||
On March 27, 2020, the Coronavirus Aid, Relief, and Economic Security Act (CARES Act) was enacted in response to the COVID-19 pandemic. The CARES Act, among other things, permits NOL carryovers and carrybacks to offset 100% of taxable income for taxable years beginning before 2021. In addition, the CARES Act allows NOLs incurred in 2018, 2019, and 2020 to be carried back to each of the five preceding taxable years to generate a refund of previously paid income taxes. The Company is currently evaluating the impact of the CARES Act, but due to sustained losses, the NOL carryback provision of the CARES Act would not yield a benefit to us.
| F-34 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and amounts used for income tax purposes. Significant components of the Company’s deferred tax assets and liabilities consist of the following:
| Year Ended | ||||||||
| June 30, 2020 | June 30, 2019 | |||||||
| Deferred tax assets | ||||||||
| Warrant Derivative Liability | $ | 7,403 | $ | 7,403 | ||||
| Accrued Expenses | 297,086 | 198,193 | ||||||
| Prepaid Investor Services | 470,050 | 414,396 | ||||||
| Non-cash interest | 596,004 | - | ||||||
| Intangibles (Intellectual Property and Patent Cost) | 240,428 | 212,881 | ||||||
| Deferred Rent | 1,969 | - | ||||||
| Formation Expense | 7,208 | 7,208 | ||||||
| Net Operating Loss carry forward | 7,438,911 | 6,573,215 | ||||||
| Foreign Exchange Loss (OCI) | (39,379 | ) | (39,379 | ) | ||||
| Revalue of derivative liability | 438,239 | 519,151 | ||||||
| Stock Based Compensation | 51,481 | 51,481 | ||||||
| Total Deferred tax assets | $ | 9,509,400 | $ | 7,944,549 | ||||
| Deferred tax liabilities | ||||||||
| R&D | $ | (177,702 | ) | $ | (139,833 | ) | ||
| Gain on extinguishment of debt | (266,987 | ) | - | |||||
| Capital Raising Costs | (255,614 | ) | (133,335 | ) | ||||
| Total deferred tax liabilities | $ | (700,303 | ) | $ | (273,168 | ) | ||
| Net deferred tax assets | $ | 8,809,097 | $ | 7,671,381 | ||||
| Valuation allowance | (8,809,097 | ) | (7,671,381 | ) | ||||
| Net deferred taxes | $ | - | $ | - | ||||
At June 30, 2020, the Company had U.S. net operating loss carry forwards of approximately $8,977,683 that may be offset against future taxable income, subject to limitation under IRC Section 382. At June 30, 2020, the Company had Australian net operating loss carry forwards of approximately $20,194,901 million which can be carried forward without expiration. No tax benefit has been reported in the June 30, 2020 and 2019 consolidated financial statements due to the uncertainty surrounding the realizability of the benefit, based on a more likely than not criteria and in consideration of available positive and negative evidence.
Management has determined that the realization of the net deferred tax asset is not assured and has created a valuation allowance for the entire amount of such benefits.
The Company follows ASC 740-10, which provides guidance for the recognition and measurement of certain tax positions in an enterprise’s financial statements. Recognition involves a determination whether it is more likely than not that a tax position will be sustained upon examination with the presumption that the tax position will be examined by the appropriate taxing authority having full knowledge of all relevant information.
The Company applied the “more-likely-than-not” recognition threshold to all tax positions taken or expected to be taken in a tax return, which resulted in no unrecognized tax benefits as of June 30, 2020 and 2019, respectively.
The Company’s policy is to record interest and penalties associated with unrecognized tax benefits as additional income taxes in the consolidated statement of operations. As of June 30, 2020, the Company had no unrecognized tax benefits. There were no changes in the Company’s unrecognized tax benefits during the years ended June 30, 2020 and 2019. The Company did not recognize any interest or penalties during fiscal 2020 or 2019 related to unrecognized tax benefits.
The income tax returns filed for the tax years from inception will be subject to examination by the relevant taxing authorities.
NOTE 8 – STOCKHOLDERS’ DEFICIT
Increase in Authorized Shares of Common Stock and Reverse Stock Split
On June 24, 2019, the Company effected a one-for-five hundred (1:500) reverse stock split whereby the Company (i) decreased the number of authorized shares of common stock, $0.001 par value per share, to 100,000,000 and (ii) decreased by a ratio of one-for-five hundred (1:500) the number of retroactively issued and outstanding shares of common stock. Proportional adjustments for the reverse stock split were made to the Company’s outstanding stock options, warrants and equity incentive plans. All share and per-share data and amounts have been retroactively adjusted as of the earliest period presented in the consolidated financial statements to reflect the reverse stock split.
On February 4, 2020 the Directors resolved to increase the Common Stock of the Company from 100,000,000 authorized shares to 1,000,000,000 authorized shares and believes that such number of authorized shares of Common Stock will be in the best interests of the Corporation and its stockholders because the Board believes that the availability of more shares of Common Stock for issuance will allow the Corporation greater flexibility in pursuing financing from investors, meeting business needs as they arise, taking advantage of favorable opportunities and responding to a changing corporate environment. The Company filed the necessary documents with the U.S. Securities and Exchange Commission on February 6, 2020 and with the amendment to the authorized shares being approved by the State of Delaware on March 13, 2020.
| F-35 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Preferred Stock
The total number of shares of preferred stock that the Company is authorized to issue is 1,500,005, $0.01 par value per share. These preferred shares have no rights to dividends, profit sharing or liquidation preferences.
Of the total preferred shares authorized, 500,000 have been designated as Series A Preferred Stock (“Series A Preferred Stock”), pursuant to the Certificate of Designation filed with the Secretary of State of the State of Delaware on December 9, 2014. James Nathanielsz, the Company’s Chief Executive Officer and Chief Financial Officer, beneficially owns all of the outstanding shares of Series A Preferred Stock via North Horizon Pty Ltd., which entitles him, as a holder of Series A Preferred Stock, to vote on all matters submitted or required to be submitted to a vote of the Company’s stockholders, except election and removal of directors, and each share of Series A Preferred Stock entitles him to two votes per share of Series A Preferred Stock. North Horizon Pty Ltd. is a Nathanielsz Family Trust. Mr. James Nathanielsz, the Chief Executive Officer, Chief Financial Officer and a director of our Company, has voting and investment power over these shares. 500,000 shares of Series A Preferred Stock are issued and outstanding as of June 30, 2020 and 2019.
Of the total preferred shares authorized, pursuant to the Certificate of Designation filed with the Secretary of State of the State of Delaware on June 16, 2015, up to five shares have been designated as Series B Preferred Stock (“Series B Preferred Stock”). Each holder of outstanding shares of Series B Preferred Stock is entitled to voting power equivalent to the number of votes equal to the total number of shares of common stock outstanding as of the record date for the determination of stockholders entitled to vote at each meeting of stockholders of the Company and entitled to vote on all matters submitted or required to be submitted to a vote of the stockholders of the Company. One share of Series B Preferred Stock is issued and outstanding as of June 30, 2020 and 2019. Mr. Nathanielsz directly beneficially owns such one share of Series B Preferred Stock.
No shares of Series A Preferred Stock or Series B Preferred Stock were issued in fiscal years 2020 or 2019.
Common Stock
Shares Issued for Cash
October 5, 2018 Equity Purchase Agreement
On October 5, 2018 (the “L2 Closing Date”), the Company entered into an Equity Purchase Agreement (the “L2 Purchase Agreement”) with L2 Capital, LLC (“L2 Capital”) pursuant to which L2 Capital committed to purchase up to $10,000,000 (the “Maximum Amount”) of the Company’s common stock (the “L2 Financing”). On the L2 Closing Date, the Company issued 7,701 shares of its common stock to L2 Capital as a commitment fee (the “Commitment Shares”), at a fair market value of $41.30 or $318,059, which was recorded as deferred offing costs and were amortized as a percentage of the Maximum Amount on a pro-rata conversion amount. Additionally, the proceeds received from the first put notice were net of $15,000 in legal fees and were recorded as deferred offering costs. Total amortization expense for the fiscal year ended June 30, 2019 was $333,059. The Commitment Shares are subject to a lock-up/leak-out limitation as described below. In connection with the L2 Financing, on the L2 Closing Date, the Company and L2 Capital also entered into a Registration Rights Agreement (the “L2 Registration Rights Agreement”, and together with the Purchase Agreement, the “L2 Transaction Documents”). The Company received net proceeds from the sale of the Put Shares directly to the Investor pursuant to the Purchase Agreement, however, the Company did not receive any proceeds from the resale of the Put Shares by L2 Capital thereafter.
Upon filing and effectiveness of the Company’s Registration Statement on Form S-1, which was declared effective by the SEC on October 30, 2018, and provided other closing conditions are met, from time to time over the term of the Purchase Agreement, the Company had the right, but not the obligation, to direct the Investor to purchase shares of the Company’s common stock (the “L2 Put Shares”) in a maximum amount of $1,000,000, provided that the number of L2 Put Shares did not exceed 250% of the Average Daily Trading Volume (as defined in the L2 Purchase Agreement). At any time and from time to time during the 3-year term of the L2 Purchase Agreement (the “Commitment Period”), the Company had the right to deliver a notice L2 Capital (the “L2 Put Notice”) and was obligated to deliver the Put Shares to Investor via DWAC (as defined in the L2 Purchase Agreement) within two trading days. The purchase price (the “L2 Purchase Price”) for the Put Shares was 87.5% of the one lowest daily volume weighted average price on the Principal Market (as defined in the L2 Purchase Agreement) (as reported by Bloomberg Finance L.P.) during the five trading days immediately following the date L2 Capital receives the L2 Put Shares via DWAC associated with the applicable Put Notice (the “L2 Valuation Period”). The closing of a Put Notice occurred within one trading day following the end of the respective L2 Valuation Period, whereby (i) L2 Capital was obligated to deliver the L2 Investment Amount (as defined below) to the Company by wire transfer of immediately available funds and (ii) L2 Capital was obligated to return surplus L2 Put Shares if the value of the L2 Put Shares delivered to L2 Capital caused the Company to exceed the maximum commitment amount. The Company could not deliver another L2 Put Notice to L2 Capital within ten trading days of a prior Put Notice. The “L2 Investment Amount” means the aggregate L2 Purchase Price for the L2 Put Shares purchased by L2 Capital, minus clearing costs due to L2 Capital’s broker or to the Company’s transfer agent for the issuance of the L2 Put Shares (the “L2 Clearing Costs”).
| F-36 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
The right of the Company to issue and sell the L2 Put Shares to L2 Capital was subject to the satisfaction of certain closing conditions, including, but not limited to, (i) the Company’s Registration Statement on Form S-1 registering for resale by the Investor of the L2 Put Shares and Commitment Shares continuing to be effective as was declared by the U.S. Securities and Exchange Commission (the “SEC”) on October 30, 2018, (ii) accuracy of the Company’s representations and warranties, (iii) the Company’s performance under the L2 Purchase Agreement in all material respects, (iv) no suspension of trading or delisting of the Company’s common stock, (v) limitation of L2 Capital’s beneficial ownership to no more than 9.99%, (vi) the Company maintaining its DWAC-eligible status, (vii) the Company maintaining a sufficient share reserve, and (viii) the minimum pricing for the L2 Put Shares must exceed $.0.05.
Pursuant to the terms of the L2 Registration Rights Agreement, the Company filed the L2 Registration Statement on October 17, 2018 and the Registration Statement was declared effective by the SEC on October 30, 2018.
L2 Capital agreed, for a period of 180 days from the L2 Closing Date, not to sell, on any given day, a number of Commitment Shares that exceeds the greater of (i) 5% of the average daily trading volume of the Company’s shares of common stock for the period ended one trading day prior to the date of such sale, as reported on the Principal Market; and (ii) such number of Commitment Shares that equals (x) $5,000, divided by, (y) the closing price of the Company’s shares of common stock one trading day prior to the date of such sale, as reported on the Principal Market.
Effective as of the L2 Closing Date, the Company reserved 924,143 shares of its common stock from its authorized and unissued shares of common stock to provide for all issuances of shares of common stock under the L2 Transaction Documents (in the event that the Company issued and sold the L2 Put Shares up to the Maximum Amount) and was required to reserve and keep available out of its authorized and unissued shares of common stock a number of shares of common stock at least three times the number of shares of common stock obtained by dividing the remaining balance on the maximum commitment amount by the L2 Purchase Price. While the Company had the obligation to maintain such reserve while the Purchase Agreement was effective, the Company did not have the obligation to sell any L2 Put Shares to L2 Capital. L2 Capital agreed, and agreed to cause any affiliate of L2 Capital acting on its behalf or pursuant to any understanding with it, not to execute any short sales during the period from the date hereof to the end of the Commitment Period.
As of about February 7, 2019, the Company reached the maximum number of shares that it could put under the L2 Purchase Agreement, and therefore, the Company would have had to file with the SEC a new registration statement registering additional shares under the L2 Purchase Agreement if the Company had determined to continue to utilize the L2 Purchase Agreement.
During the fiscal year ended June 30, 2019, the Company issued 113,200 shares of its common stock at an average price per share of $8.50, ranging from $6.50 to $20.00, as a result of delivering 14 L2 Put Notices to L2 Capital. As of June 30, 2019, the Company received gross aggregate proceeds of $964,009 from such put notices. Effective as of February 25, 2019, the Company terminated the L2 Purchase Agreement.
February 25, 2019 Equity Purchase Agreement
On February 25, 2019 (the “Closing Date”), the Company entered into an Equity Purchase Agreement (the “Purchase Agreement”) with a certain institutional investor (the “Investor”) pursuant to which the Investor committed to purchase up to $10,000,000 (the “Maximum Amount”) of the Company’s common stock (the “Financing”). In connection with the Financing, on the Closing Date, the Company and the Investor also entered into a Registration Rights Agreement (the “Registration Rights Agreement”, and together with the Purchase Agreement, the “Transaction Documents”). The Company will receive net proceeds from the sale of the Put Shares directly to the Investor pursuant to the Purchase Agreement, however, the Company will not receive any proceeds from the resale of the Put Shares by the Investor thereafter.
Upon filing and effectiveness of the Company’s Registration Statement on Form S-1, which was declared effective by the SEC on March 7, 2019, and provided other closing conditions are met, from time to time over the term of the Purchase Agreement, the Company has the right, but not the obligation, to direct the Investor to purchase shares of the Company’s common stock (the “Put Shares”) in a maximum amount of $1,000,000, provided that the number of Put Shares did not exceed 250% of the Average Daily Trading Volume (as defined in the Purchase Agreement). At any time and from time to time during the 3-year term of the Purchase Agreement (the “Commitment Period”), the Company has the right to deliver a notice to the Investor (the “Put Notice”) and is obligated to deliver the Put Shares to Investor via DWAC (as defined in the Purchase Agreement) within two trading days. The purchase price (the “Purchase Price”) for the Put Shares was 87.5% of the one lowest daily volume weighted average price on the Principal Market (as defined in the Purchase Agreement) (as reported by Bloomberg Finance L.P.) during the five trading days immediately following the date the Investor receives the Put Shares via DWAC associated with the applicable Put Notice (the “Valuation Period”). The closing of a Put Notice occurs within one trading day following the end of the respective Valuation Period, whereby (i) the Investor is obligated to deliver the Investment Amount (as defined below) to the Company by wire transfer of immediately available funds and (ii) the Investor is obligated to return surplus Put Shares if the value of the Put Shares delivered to the Investor causes the Company to exceed the maximum commitment amount. The Company cannot deliver another Put Notice to the Investor within ten trading days of a prior Put Notice. The “Investment Amount” means the aggregate Purchase Price for the Put Shares purchased by the Investor, minus clearing costs due to the Investor’s broker or to the Company’s transfer agent for the issuance of the Put Shares (the “Clearing Costs”).
| F-37 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
The right of the Company to issue and sell the Put Shares to the Investor is subject to the satisfaction of certain closing conditions, including, but not limited to, (i) the Company’s Registration Statement on Form S-1 registering for resale by the Investor of the Put Shares continuing to be effective as was declared by the SEC on March 7, 2019, (ii) accuracy of the Company’s representations and warranties, (iii) the Company’s performance under the Purchase Agreement in all material respects, (iv) no suspension of trading or delisting of the Company’s common stock, (v) limitation of the Investor’s beneficial ownership to no more than 9.99%, (vi) the Company maintaining its DWAC-eligible status, (vii) the Company maintaining a sufficient share reserve, and (viii) the minimum pricing for the Put Shares must exceed $0.05.
Pursuant to the terms of the Registration Rights Agreement, the Company filed the Registration Statement on February 25, 2019 and the Registration Statement was declared effective by the SEC on March 7, 2019.
Effective as of the Closing Date, the Company reserved 1,333,333 shares of its common stock from its authorized and unissued shares of common stock to provide for all issuances of shares of common stock under the Transaction Documents (in the event that the Company issued and sold the Put Shares up to the Maximum Amount) and was required to reserve and keep available out of its authorized and unissued shares of common stock a number of shares of common stock at least three times the number of shares of common stock obtained by dividing the remaining balance on the maximum commitment amount by the Purchase Price.
While the Company has the obligation to maintain such reserve while the Purchase Agreement was effective, the Company does not have the obligation to sell any Put Shares to the Investor. The Investor agreed, and agreed to cause any affiliate of the Investor acting on its behalf or pursuant to any understanding with it, not to execute any short sales during the period from the date hereof to the end of the Commitment Period.
During the year ended June 30, 2019, the Company issued 34,000 shares of its common stock at an average price per share of $4.01, ranging from $3.06 to $4.90, as a result of delivering five Put Notices to the Investor. As of June 30, 2019, the Company received gross aggregate proceeds of $136,371 from such put notices.
April 3, 2020 Security Purchase Agreement
On April 3, 2020, the Company closed on a transaction related to a Securities Purchase Agreement (the “Securities Purchase Agreement”) entered into on March 30, 2020, whereby an investor (the “Investor”) purchased from the Company, 7,500,000 units (the “Units”), each consisting of (i) 1.5 shares of the Company’s common stock (the “Common Stock”), or pre-funded warrants (the “Prefunded Warrants”) upon Investor’s election due to the 4.99% blocker provision as discussed below and (ii) 1.5 warrants to purchase one share of Common Stock (“Series A Warrants”), along with such purchaser’s pro-rata portion of the Series B Warrants and Series C Warrants (“the Units”). In aggregate the Investor was issued 63,750,000 warrants to purchase one share of Common Stock (the “Series B Warrants”) and an additional 63,750,000 warrants to purchase one share of Common Stock, subject to a vesting schedule based on the Investors exercise of the Series B Warrants (the “Series C Warrants” and, together with the Prefunded Warrants, the Series A Warrants, and the Series B Warrants referred to herein as, the “Warrants”). See discussion of warrant terms under “Warrants” below.
The aggregate purchase price for the Units, the Series A Warrants with exercise price of $0.20 per share, the Series B Warrants with exercise price of $0.04 per share and the Series C Warrants with exercise price of $0.20 per share, of $450,000 was paid at closing (the “Purchase Price”) or $0.06 per unit purchase price (see Warrants below). The Company received net proceeds of $424,990, net of offering cost of $25,010.
The Securities Purchase Agreement contains a blocker provision whereby the Investor or any of its affiliates would not beneficially own in excess of 4.99% of the outstanding number of shares of Common Stock (“Beneficial Ownership Limitation”). As such, the Investor may elect to purchase Prefunded Warrants equal to the same number of shares of Common Stock that the Company would have been issued.
Due to the Beneficial Ownership Limitation, the 11,250,000 shares of Common Stock underlying the Units issuable at closing of the Securities Purchase Agreement are comprised of 804,518 shares of restricted Common Stock and 10,445,482 Prefunded Warrants with exercise price of $0.0001 (but can be less than par value). The Prefunded Warrants shall be exercisable immediately and shall expire when exercised in full.
The Securities Purchase Agreement contains such representations, warranties and covenants as are typical for a transaction of this nature.
Shares issued for conversion of convertible debt
During the year ended June 30, 2019, the Company issued 704,258 shares of its common stock at an average contractual conversion price of $4.76, ranging from approximately $0.61 to $14.97, as a result of the conversion of principal and interest in the aggregate amount of $3,350,783 underlying certain outstanding convertible notes converted during such period. The total recorded to equity was $3,350,783. The Company reclassified $1,824,317 in put premiums to additional paid in capital following conversions during the year ended June 30, 2019.
| F-38 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
During the year ended June 30, 2020, the Company issued 247,619,247 shares of its common stock at an average contractual conversion price of $0.007, ranging from $0.002 to $0.91, as a result of the conversion of principal and interest in the aggregate amount of $1,814,336 underlying certain outstanding principal amount and accrued interest of convertible notes converted during such period, including $15,000 of conversion fees. The total recorded to equity was $2,125,174. Notes with principal amounts totaling $254,500 and accrued interest of $15,408 contained bifurcated embedded conversion option derivatives. Accordingly, the fair market value of the shares issued was $565,746 resulting in a loss on extinguishment at the time of conversion of $295,838 and $362,961 of derivative fair value was recorded as a gain on extinguishment at the time of conversion. The Company reclassified $874,924 in put premiums to additional paid in capital following conversions during the year ended June 30, 2020.
The Company has 739,138,743 shares of its common stock reserved for future issuances based on lender reserve requirements pursuant to underlying financing agreements at June 30, 2020.
Shares issued for services
On December 6, 2018, the Company entered into an agreement with a certain consultant to provide services over a six-month period beginning November 1, 2018 and ending May 1, 2019 in exchange for 4,000 shares of the Company’s common stock. On December 27, 2018, the Company issued the 4,000 shares of the Company’s common stock valued at $10.00 per share to such consultant, or $39,000, which will be amortized over the term of the agreement. The Company recorded $39,000 of consulting expense with respect to such shares of its common stock during the year ended June 30, 2019.
On November 20, 2018, the Company’s Board of Directors authorized the issuance of 2,000 shares of the Company’s common stock in connection with certain legal services provided to the Company. On November 28, 2018, the Company issued such 2,000 shares of its common stock valued at $15.00, or $30,000.
In March 2019 and effective as of December 21, 2018, the Company entered into a certain consulting services agreement with a certain consultant to provide services over a twelve-month period beginning December 21, 2018 in exchange for issuance of two tranches of 10,000 shares (subject to certain true-up provisions), for services to be rendered between December 21, 2018 and March 20, 2019, and 6,000 shares (subject to certain true-up provisions), for services to be rendered between March 21, 2019 and December 20, 2019 of the Company’s common stock. On May 8, 2019, the Company terminated the agreement with the consent of the consultant. The consultant agreed that the issuance of the first tranche of 10,000 shares (including the true-up provision) together with cash payments already made by the Company to the consultant fully satisfied the obligations (past and future) that the Company has under the consulting agreement including any claims under the true-up provisions of the agreement. In March 2019, the Company issued the first tranche of 10,000 shares of its common stock valued at $10.00 per share based on the quoted trading price to the consultant, or $100,000. The Company recorded $100,000 of consulting expense with respect to such shares of its common stock during the fiscal year ended June 30, 2019.
On July 19, 2019, the Company entered into an agreement with a certain consultant to provide services over a two-month period beginning July 1, 2019 and ending September 1, 2019 in exchange for 20,000 shares of the Company’s common stock. On July 19, 2019, the Company issued the 20,000 shares of the Company’s common stock valued at $1.99 per share; being the closing price of the stock on the date of the agreement, to such consultant, or $39,800, which will be amortized over the term of the agreement. The Company recorded $39,800 of consulting expense with respect to such shares of its common stock during the year ended June 30, 2020.
Between February 3, 2020 and June 26, 2020, the Company issued an aggregate of 8,708,574 shares of the Company’s common stock to a consultant for services rendered pursuant to an engagement agreement dated on September 10, 2019 which agreement was later amended in February 2020. Between February 3, 2020 and June 26, 2020, the Company issued an aggregate of 8,708,574 shares of the Company’s common stock valued at an average price of $0.008 per share; being the closing price of the stock on the date of the agreement, to such consultant, or $73,842. The Company recorded $73,842 of consulting expense with respect to such shares of its common stock during the year ended June 30, 2020.
Restricted Stock Units
Pursuant to employment agreements dated in May 2019 (see Note 9), the Company granted an aggregate of 78,000 and 39,000 restricted stock unit to the Company’s Chief Executive Officer and Chief Scientific Officer, respectively. The total 117,000 restricted stock units are subject to vesting terms as defined in the employment agreements. The 117,000 restricted stock units were valued at the fair value of $4.25 per unit or $497,240 based on the quoted trading price on the date of grant. During the year ended June 30, 2020 and 2019, the Company recognized stock-based compensation of $217,543 and $31,077, respectively, related to vested restricted stock units. There were $248,620 unrecognized restricted stock units expense as of June 30, 2020 which may be recognized upon achievement of certain performance conditions.
| F-39 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Stock Options
On May 14, 2019, the Company’s board of directors approved and adopted the Company’s 2019 Equity Incentive Plan (the “2019 Plan”), which reserves a total of 234,000 shares of the Company’s common stock for issuance under the 2019 Plan. Incentive awards authorized under the 2019 Plan include, but are not limited to, incentive stock options, non-qualified stock options, restricted stock awards and restricted stock units.
Pursuant to employment agreements dated in May 2019 (see Note 10), the Company granted options to purchase 39,000 and 19,500 shares of the Company’s common stock to the Company’s Chief Executive Officer and Chief Scientific Officer, respectively. The total 58,500 options have a term of 10 years from the date of grant and exercise price ranging from $4.25 to $4.675 per share. 1/3rd of these options shall vest every successive one-year anniversary, provided, that on each such vesting date, the Chief Executive Officer and Chief Scientific Officer are employed by the Company and subject to the other provisions of the employment agreement. The 58,500 stock options were valued using a Black-Scholes model with the following assumptions: stock price at valuation date of $4.25 based on quoted trading price on date of grant, exercise price of $4.65, dividend yield of zero, years to maturity of 10.00, a risk free rate of 2.42%, and expected volatility 268% for a total value of $248,620.
During the year ended June 30, 2020 and 2019, the Company recognized stock-based compensation of $82,873 and $10,360 related to vested stock options. There was $155,387 of unvested stock options expense as of June 30, 2020 that will be recognized through May 2022 or 1.85 years.
A summary of the Company’s option activity during the years ended June 30, 2020 and 2019 is presented below:
| Weighted | ||||||||
| Number of | Average Exercise | |||||||
| Shares | Price Per Share | |||||||
| Outstanding at June 30, 2018 | 1,144 | $ | 3,750 | |||||
| Issued | 58,500 | 4.53 | ||||||
| Exercised | - | - | ||||||
| Expired | - | - | ||||||
| Outstanding at June 30, 2019 | 59,644 | $ | 76.37 | |||||
| Issued | - | - | ||||||
| Exercised | - | - | ||||||
| Forfeited | - | - | ||||||
| Expired | - | - | ||||||
| Outstanding at June 30, 2020 | 59,644 | $ | 76.37 | |||||
| Exercisable at June 30, 2020 | 20,644 | $ | 212.09 | |||||
| Outstanding and Exercisable: | ||||||||
| Weighted average remaining contractual term | 8.72 | |||||||
| Weighted average fair value of options granted during the period | $ | - | ||||||
| Aggregate intrinsic value | $ | - | ||||||
| Options Outstanding | Options Exercisable | ||||||||||||||||||||||||||||||
Range of Exercise Price | Number Outstanding at June 30, 2020 | Weighted Average Remaining Contractual Life | Weighted Average Exercise Price | Aggregate Intrinsic Value | Number Exercisable at June 30, 2020 | Weighted Average Exercise Price | Aggregate Intrinsic Value | ||||||||||||||||||||||||
| $ | 4.25-4.68 | 58,500 | 8.88 Years | $ | 4.53 | $ | - | 19,500 | $ | 4.50 | $ | - | |||||||||||||||||||
| $ | 3,750.00 | 1,144 | 0.79 Years | 3,750.00 | - | 1,144 | 3,750.00 | - | |||||||||||||||||||||||
| 59,644 | 8.72 Years | $ | 76.37 | $ | - | 20,644 | $ | 212.09 | $ | - | |||||||||||||||||||||
Warrants
On August 29, 2018, the Company received payment of $39 AUD for the exercise of a warrant for 24 shares of the Company’s common stock and issued such shares as a result of the exercise.
On December 2, 2018, a total of 208 warrants expired.
| F-40 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
No warrants were issued during the year ended June 30, 2019.
In connection with the issuance of the August 2019 Auctus Note, the Company issued common stock purchase warrants to Auctus to purchase 450,000 shares of the Company’s common stock (the “First Warrant”) as a commitment fee upon the terms and subject to the limitations and conditions set forth in such First Warrant at an “Exercise Price” of $2.25. In connection with the issuance of the Note, the Company shall issue a common stock purchase warrant to Buyer to purchase 300,000 shares of the Company’s common stock (the “Second Warrant”) as a commitment fee upon the terms and subject to the limitations and conditions set forth in such Second Warrant at an “Exercise Price” of $3.33. In connection with the issuance of the Note, the Company shall issue a common stock purchase warrant to Buyer to purchase 225,000 shares of the Company’s common stock (the “Third Warrant”) as a commitment fee upon the terms and subject to the limitations and conditions set forth in such Third Warrant at an “Exercise Price” of $4.50. The First Warrant, Second Warrant, and Third Warrant shall collectively be referred as the “Warrants”. The Warrants have an “Exercise Period” of five years form the date of issuance being August 30, 2019 (see Note 6).
On September 10, 2019, the Company entered into an agreement with a certain consultant to provide services over a three-month period beginning September 10, 2019 and ending December 10, 2019 in exchange for 1,000,000 warrants to purchase the Company’s common stock at $2.00 per share with an expiry date of September 10, 2022. The Fair Market Value of the warrants was $984,810 on the date of grant as calculated under the Black Scholes Option Pricing model. The Company recorded $984,810 of share-based compensation expenses with respect to the grant of such warrants during the year ended June 30, 2020.
In connection with the issuance of shares on April 3, 2020 as discussed above, the Company closed on a transaction related to a Securities Purchase Agreement (the “Securities Purchase Agreement”) entered into on March 30, 2020, whereby an investor purchased from the Company, 7,500,000 units, each consisting of (i) 1.5 shares of the Company’s common stock, or pre-funded warrants upon Investor’s election due to the 4.99% blocker provision and (ii) 1.5 warrants to purchase one share of Common Stock (“Series A Warrants”, and collectively with the Common Stock the “Units”). In addition to the Units, the Investor was issued 63,750,000 warrants to purchase one share of Common Stock (the “Series B Warrants”) and an additional 63,750,000 warrants to purchase one share of Common Stock, subject to a vesting schedule (the “Series C Warrants” and, together with the Prefunded Warrants, the Series A Warrants, and the Series B Warrants, the “Warrants”).
Due to the Beneficial Ownership Limitation, the Company granted 10,445,482 Prefunded Warrants with exercise price of $0.0001 (but can be less than par value). The Prefunded Warrants shall be exercisable immediately and shall expire when exercised in full. On July 22, 2020, the Company received proceeds of $1,045 from the exercise of 10,445,482 Prefunded Warrants (see Note 13).
Series A Warrants
As discussed above, pursuant to the Securities Purchase Agreement entered into March 20, 2020, the Investor purchased Series A Warrants to purchase up to 11,250,000 shares of Common Stock, subject to adjustment as provided therein. The Series A Warrants have a cash exercise price of $0.20 per share and are immediately exercisable and expire in 3 years. The Series A Warrants contain a provision for cashless exercise in the event there is no effective registration statement registering the shares underlying the Series A Warrants calculated based on the difference between the exercise price of the Series A Warrant and the trading price of the stock (the “Cashless Exercise”).Additionally, the Series A Warrants contain another provision for a cashless exercise at the Holder’s option should the trading price of the Common Stock fall below $0.20 per share calculated based on the difference between the exercise price of the Series A Warrant and 70% of the Market Price, as defined therein (the” Alternate Cashless Exercise”).
Series B Warrants
As discussed above, pursuant to the Securities Purchase Agreement entered into March 20, 2020, the Investor purchased Series B Warrants to purchase up to 63,750,000 shares of Common Stock, subject to adjustment as provided therein; provided, however, commencing on the 90th day following the effective date, the Company may reduce the number of Warrant Shares issuable upon exercise thereof by 37,500,000 upon 10 Trading Days’ prior written notice to the Holder provided that the Company issues to the Holder 3,750,000 shares of Common Stock (or, at the election of the Holder, an equivalent number of pre-funded warrants) and Series A Warrants to purchase up to 3,750,000 shares of Common Stock, which shares shall be issued pursuant to a registration statement without restrictions on resale. The Series B Warrants have a cash exercise price of $0.04 per share and expire in 3 years. The Series B Warrants contain a provision for Cashless Exercise. On July 22, 2020, the Company received proceeds of $100,000 from the exercise of 2,500,000 Series B Warrants (see Note 13).
Series C Warrants
As discussed above, pursuant to the Securities Purchase Agreement entered into March 20, 2020, the Investor purchased Series C Warrants to purchase up to 63,750,000 shares of Common Stock, subject to adjustment as provided therein and expire in 3 years. The Series C Warrants have a cash exercise price of $0.20 per share, subject to a vesting schedule, which is based on such Holder’s exercise of the Series B Warrants. The Series C Warrants contain provisions for Cashless Exercise and Alternate Cashless Exercise.
| F-41 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
The following table summarizes warrant activity for the years ended June 30, 2020 and 2019:
| Weighted | ||||||||
| Number of | Average | |||||||
| Shares | Price Per Share | |||||||
| Outstanding at June 30, 2018 | 291 | $ | 5,555 | |||||
| Issued | - | - | ||||||
| Exercised | (24 | ) | - | |||||
| Forfeited | - | - | ||||||
| Expired | (208 | ) | - | |||||
| Outstanding at June 30, 2019 | 59 | $ | 4,765 | |||||
| Issued | 151,170,482 | 0.15 | ||||||
| Exercised | - | - | ||||||
| Forfeited | - | - | ||||||
| Expired | (27 | ) | - | |||||
| Outstanding at June 30, 2020 | 151,170,514 | $ | 0.15 | |||||
| Exercisable at June 30, 2020 | 151,170,482 | $ | 0.15 | |||||
| Outstanding and Exercisable: | ||||||||
| Weighted average remaining contractual term | 2.76 | |||||||
| Aggregate intrinsic value | $ | 30,292 | ||||||
NOTE 9 – COMMITMENTS AND CONTINGENCIES
Legal Matters
A complaint against us, dated September 26, 2019, has been filed by Foley Shechter Ablovatskiy (“Foley Shechter”), our former counsel, seeking $151,031.50 in legal fees, in addition to interest and costs of suit. The Company believes these claims to be unfounded and is vigorously defending itself. To that end, on November 20, 2019 the Company filed a motion to dismiss certain counts of the complaint, with prejudice. That motion remains pending with the Supreme Court of the State of New York, County of New York. Oral argument is scheduled for November 5, 2020. Upon resolution of the motion, the Company shall file an answer, together with affirmative defences and counterclaims. The counterclaims shall include, without limitation, malpractice claims, arising out of Foley Shechter’s grossly negligent mishandling of certain transactions and excessive billing related thereto. Certain amounts related to this claim are included in accounts payable and accrued expenses in the accompanying Financial Statements. If our motion to dismiss is granted, our potential liability would be reduced to $51,031.51 plus interest and attorney’s fees.
Regal Consulting, LLC (“Regal”) initiated litigation against the Company in Clark County District Court, Nevada. Regal is demanding approximately $400,000 and 60,000 shares of the Company’s common stock as payment for services that Regal purports to have performed. Regal additionally claims that $106,500 remains due on a Convertible Note executed by the Company in May of 2017 (the “2017 Note”), and asserts that it is owed in excess of $100,000 in penalties in connection with the Company’s refusal to honor certain Conversion Notices. The Company filed an Answer and Counterclaim, denying liability and alleging that Regal procured by fraud the Company’s execution of various consulting agreements and additionally failed to provide the consulting services contemplated by said agreements.
The discovery process is ongoing. In addition, the parties have agreed to mediate their dispute and are in the process of selecting a mediator and scheduling their mediation.
IRS Liability
As part of its requirement for having a foreign operating subsidiary, the Company’s parent U.S. entity is required to file an informational Form 5471 to the Internal Revenue Service (the “IRS”), which is a form that explains the nature of the relationship between the foreign subsidiary and the parent company. From 2012 through the 2014, the Company did not file this form in a timely manner. As a result of the non-timely filings, the Company incurred a penalty from the IRS in the amount of $10,000 per year, or $30,000 in total, plus accrued interest, such penalty and interest having been accrued and is included in the Accrued expenses and other payable figure in the consolidated balance sheet. The Company recorded the penalties for all three years during the year ended June 30, 2018 and is negotiating a payment plan. The Company is current on all subsequent filings.
| F-42 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Operating Agreements
In November 2009, the Company entered into a commercialization agreement with the University of Bath (UK) (the “University”) whereby the Company and the University co-owned the intellectual property relating to the Company’s pro-enzyme formulations. In June 2012, the Company and the University entered into an assignment and amendment whereby the Company assumed full ownership of the intellectual property while agreeing to pay royalties of 2% of net revenues to the University. Additionally, the Company agreed to pay 5% of each and every license agreement subscribed for. The contract is cancellable at any time by either party. To date, no amounts are owed under the agreement.
Operating Leases
On May 5, 2016, the Company entered into a new five-year operating lease agreement with a Horizon Pty Ltd., a related party, of which Mr. Nathanielsz, our CEO, CFO and a director, and his wife are owners and directors, with monthly rent currently at $3,606 AUD or $2,431 USD, inclusive of GST (See Note 10 – Related Party Transactions). The initial rental amount was $3,000 AUD subject to a 3% yearly escalation. In adopting ASC Topic 842, Leases (Topic 842), the Company has elected the ‘package of practical expedients’, which permit it not to reassess under the new standard its prior conclusions about lease identification, lease classification and initial direct costs. In addition, the Company elected not to apply ASC Topic 842 to arrangements with lease terms of 12 month or less. On July 1, 2019, upon adoption of ASC Topic 842, the Company recorded right-of-use assets $48,662 and total lease liabilities of $48,662 based on an incremental borrowing rate of 6%.
ROU is summarized below:
| June 30, 2020 | ||||
| Office lease ROU | $ | 48,662 | ||
| Less accumulated reduction | (26,980 | ) | ||
| Balance of ROU asset as of June 30, 2020 | $ | 21,682 | ||
Operating lease liability related to the ROU asset is summarized below:
| June 30, 2020 | ||||
| Office lease liability | $ | 48,662 | ||
| Reduction of lease liability | (23,590 | ) | ||
| Total | $ | 25,072 | ||
Future Minimum lease payments under non-cancelable operating lease at June 30, 2020 are as follows:
| Fiscal Year 2021 | $ | 25,700 | ||
| Imputed interest | (628 | ) | ||
| Total operating lease liability | $ | 25,072 |
Amatsigroup Agreement
The Company entered into a Manufacturing Services Agreement (the “MSA”) and Quality Assurance Agreement (the “QAA”), each with an effective date of August 12, 2016, with Amatsigroup NV (“Amatsigroup”), formerly known as Q-Biologicals, NV, a contract manufacturing organization located in Belgium. Pursuant to the MSA, Amatsigroup produces certain drug substances and products containing certain enzymes for the Company at its facility in Belgium. The Company uses these substances and products for development purposes, including but not limited to future clinical trials. The MSA contemplates payment to Amatsigroup pursuant to a pre-determined fee schedule based on the completion of certain milestones that depend on our manufacturing requirements and final batch yield. The Company anticipates that its payments to Amatsigroup under the MSA will range between $2.5 million and $5.0 million over three years, when the finished drug product is manufactured and released for clinical trials. The Company has spent a total of $1,689,146 of costs to date under this contract of which $49,854 was expensed in fiscal 2019, $701,973 in fiscal 2018 and $937,319 in fiscal 2017. The MSA expired in 2019 and may be extended by mutual agreement in writing with a possible extension currently under consideration. The Company can terminate the MSA early for any reason upon the required notice period, however, in such event, the pre-payment paid upon signing the MSA is considered non-refundable. Each party to the MSA shall have the right to terminate the MSA by written notice to the other party if the other party commits a material breach of the MSA (subject to a 30-day cure period). The QAA sets forth the parties’ respective obligations and responsibilities relating to the manufacturing and testing of the products under the MSA. The agreements with Amatsigroup contain certain customary representations, warranties and limitations of liabilities, and confidentiality and indemnity obligations. The MSA expired in 2019 and may be extended by mutual agreement in writing with a possible extension currently under consideration.
| F-43 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Collaboration Agreement
On September 13, 2018, the Company entered into a two-year collaboration agreement with the University of Jaén (the “University”) to provide certain research services to the Company, which agreement will be extended. In consideration of such services, the Company agreed to pay the University approximately 52,000 Euros ($59,508 USD) in year one and a maximum of 40,000 Euros ($45,775 USD) in year two. The Company paid 31,754 Euros ($36,117 USD) in 2019 and has accrued 15,410 Euros ($17,331 USD) in the year ended June 30,2020. Additionally, in exchange for full ownership of the intellectual property the Company agreed to pay royalties of 2% of net revenues to the University.
NOTE 10 – RELATED PARTY TRANSACTIONS
Since its inception, the Company has conducted transactions with its directors and entities related to such directors. These transactions have included the following:
As of June 30, 2020 and 2019, the Company owed a current and a former director a total of $50,993 and $51,867, respectively, for money loaned to the Company throughout the years. The total loans balance owed at June 30, 2020 and 2019 is not interest bearing (See Note 5 – Loans and Notes Payable).
As of June 30 2020, and 2019, the Company owed its former director a total of $30,639 and $31,164, respectively, related to expenses paid on behalf of the Company related to corporate startup costs and intellectual property (See Note 4 – Due to Former Director – Related Party).
Effective May 5, 2016, the Company entered into an agreement for the lease of its principal executive offices with North Horizon Pty Ltd., a related party, of which Mr. Nathanielsz, our CEO, CFO and a director, and his wife are owners and directors. The lease has a five-year term expiring May 2021 and provides for annual rental payments of $39,600 AUD or $28,325 USD, which includes $3,600 AUD or $2,575 USD of goods and service tax for total payments of $198,000 AUD or $141,629 USD during the term of the lease. As of June 30, 2020, total payments of $37,295 AUD or $25,700 USD remain on the lease. (See Note 9 – Commitments and Contingencies)
The Company and Mr. Nathanielsz entered into an employment agreement as of February 25, 2015 (the “Nathanielsz Employment Agreement”) setting forth the terms and conditions of Mr. Nathanielsz employment as the Company’s President and Chief Executive Officer. The Nathanielsz Employment Agreement was scheduled to expire on February 25, 2019; however, the term of the Nathanielsz Employment Agreement automatically renews for successive one-year periods unless either party provides 30 days’ prior written notice of its intent not to renew. The Nathanielsz Employment Agreement continues in effect as of June 30, 2020 as amended May 14, 2019 (see below). The Nathanielsz Employment Agreement provides Mr. Nathanielsz with a base salary of $25,000 AUD per month ($300,000 AUD annually or $205,680 USD) and a monthly contribution to Mr. Nathanielsz’s pension equal to 9.5% of his monthly salary. Mr. Nathanielsz has the ability to convert any accrued but unpaid salary into common stock at the end of each fiscal year at a conversion price to be determined by Mr. Nathanielsz and the Company, which will in no event be lower than par value or higher than the closing bid price on the date of conversion. Pursuant to the Nathanielsz Employment Agreement, Mr. Nathanielsz is entitled to an annual discretionary bonus in an amount up to 200% of his annual base salary, which bonus shall be determined by the Company’s board of directors based upon the performance of the Company. On March 16, 2018, the Company’s board of directors approved an increase of Mr. Nathanielsz’s annual base salary from $300,000 AUD ($205,680 USD) to $400,000 AUD ($274,240 USD), effective February 2018.
Mr. Nathanielsz’s wife, Sylvia Nathanielsz, is and has been a non-executive part-time employee of the Company since October 2015. Effective February 1, 2018. Mrs. Nathanielsz receives an annual salary of $120,000 AUD ($80,904 USD) and is entitled to customary benefits.
| F-44 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Pursuant to a February 25, 2016 board resolution, James Nathanielsz shall be paid $4,481 AUD ($3,205 USD), on a monthly basis for the purpose of acquiring and maintaining an automobile. For the ended June 30, 2020, a total of $44,918 AUD ($30,284 USD) in payments have been made with respect to Mr. Nathanielsz’s car allowance. For the fiscal year ended June 30, 2019, a total of $53,772 AUD ($38,463 USD) in costs have been incurred with respect to Mr. Nathanielsz’s car allowance.
Pursuant to the approval of the Company’s board of directors, on March 16, 2018, Mr. Nathanielsz was granted a $300,000 AUD ($210,090 USD) bonus for accomplishments achieved while serving as the Company’s Chief Executive Officer during the fiscal year 2018. A total of $80,046 AUD ($56,056 USD) in payments were made in fiscal 2018. During the year ended June 30, 2019, an additional $219,954 AUD ($150,602 USD) was paid. Such bonus was fully paid to Mr. Nathanielsz as of June 30, 2019.
Pursuant to the approval of the Company’s board of directors, on May 14, 2019, Mr. Nathanielsz was granted a $460,000 AUD ($315,376 USD) bonus for accomplishments achieved while serving as the Company’s Chief Executive Officer during the fiscal year ended June 30, 2019 with $200,000 AUD ($137,120 USD) of such bonus payable by the Corporation to the CEO throughout the Corporation’s 2019 fiscal year as the Corporation’s cash resources allow, with the remaining $260,000 AUD ($178,256 USD) of such bonus to be deferred by the CEO until a future date when the Corporation’s cash resources allow for such payment, as agreed to by the CEO. A total of $90,000 AUD ($64,377 USD) in payments were made in the year ended June 30, 2019. On July 13, 2020 the Board approved a bonus of $240,000 AUD being equal to 60% of Mr Nathanielsz base salary which was accrued as of June 30, 2020. A total of $202,620 AUD ($136,606 USD) in payments were made in during the year ended June 30, 2020, with $407,380 AUD ($280,726 USD) remaining due and payable.
New Employment and Services Agreements with Management
Amended and Restated Employment Agreement ― On May 14, 2019 (the “Effective Date”), the Company entered into an Amended and Restated Employment Agreement (the “Employment Agreement”) with James Nathanielsz, the Company’s Chief Executive Officer, Chairman, acting Chief Financial Officer and a director, for a term of three years, subject to automatic one-year renewals, at an annual salary of $400,000 AUD. Pursuant to the Employment Agreement, Mr. Nathanielsz was granted options to purchase 39,000 shares of the Company’s common stock (the “Nathanielsz Options”), with an exercise price per share of $4.675 (110% of the closing market price of the Company’s common stock on May 14, 2019 (or $4.25), the date of approval of such grant by the Company’s board of directors), (ii) 39,000 restricted stock units of the Company (the “Initial Nathanielsz RSUs”), and (iii) an additional 39,000 restricted stock units of the Company (the “Additional Nathanielsz RSUs”). Such options and restricted stock units were granted pursuant to the 2019 Plan approved by the Company’s board of directors on the Effective Date. The Nathanielsz Options have a term of 10 years from the date of grant. 1/3rd of the Nathanielsz Options shall vest every successive one-year anniversary following the Effective Date, provided, that on each such vesting date Mr. Nathanielsz is employed by the Company and subject to the other provisions of the Employment Agreement. The Initial Nathanielsz RSUs shall vest on the one-year anniversary of the Effective Date, subject to Mr. Nathanielsz’s continued employment with the Company through such vesting date. The Additional Nathanielsz RSUs will vest as follows, subject to Mr. Nathanielsz’s continued employment with the Company through the applicable vesting date: (i) 7,800 of the Additional Nathanielsz RSUs shall vest upon the Company submitting Clinical Trial Application (the “CTA”) for PRP, the Company’s lead product candidate (“PRP”), for a First-In-Human study for PRP (the “Study”) in an applicable jurisdiction to be selected by the Company, (ii) 7,800 of the Additional Nathanielsz RSUs shall vest upon the CTA being approved in an applicable jurisdiction, (iii) 7,800 of the Additional RSUs shall vest upon the Company completing an equity financing in the amount of at least $4,000,000 in gross proceeds, (iv) 7,800 of the Additional Nathanielsz RSUs shall vest upon the shares of the Company’s Common Stock being listed on a senior stock exchange (NYSE, NYSEMKT or NASDAQ), and (v) the remaining 7,800 of the Additional Nathanielsz RSUs shall vest upon the Company enrolling its first patient in the Study. Each vested restricted stock unit shall be settled by delivery to Mr. Nathanielsz of one share of the Company’s common stock and/or the fair market value of one share of common stock in cash, at the sole discretion of the Company’s board of directors and subject to the 2019 Plan, on the first to occur of: (i) the date of a Change of Control (as defined in the Employment Agreement), (ii) the date that is ten business days following the vesting of such restricted stock unit, (iii) the date of Mr. Nathanielsz’s death or Disability (as defined in the Employment Agreement), and (iv) Mr. Nathanielsz’s employment being terminated either by the Company without Cause or by Mr. Nathanielsz for Good Reason (each as defined in the Employment Agreement). In the event of a Change of Control, any unvested portion of the Nathanielsz Options and such restricted stock units shall vest immediately prior to such event.
The 78,000 restricted stock units were valued at the fair value of $4.25 per unit or $331,500 based on the quoted trading price on the date of grant. The 39,000 stock options were valued using a Black-Scholes model with the following assumptions: stock price at valuation date of $4.25 based on quoted trading price on date of grant, exercise price of $4.65, dividend yield of zero, years to maturity of 10.00, a risk free rate of 2.42%, and expected volatility 268% for a total value of $165,747 (see Note 8 – Stockholders’ Deficit).
| F-45 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Amended and Restated Services Agreement ― On the Effective Date, the Company also entered into an Amended and Restated Services Agreement (the “Services Agreement”) with Dr. Kenyon, the Company’s Chief Scientific Officer and a director, for a term of three years, subject to automatic one-year renewals, at an annual salary of $54,000 AUD. In connection with the execution of the Services Agreement, Dr. Kenyon was designated as an executive officer of the Company and assumed a more active executive role with the Company. Pursuant to the Services Agreement, Dr. Kenyon was granted options to purchase 19,500 shares of the Company’s common stock (the “Kenyon Options”), with an exercise price per share of $4.25 (100% of the closing market price of the Company’s common stock on May 14, 2019, the date of approval of such grant by the Company’s board of directors), (ii) 19,500 restricted stock units of the Company (the “Initial Kenyon RSUs”), and (iii) an additional 19,500 restricted stock units of the Company (the “Additional Kenyon RSUs”). Such options and restricted stock units were granted pursuant to the 2019 Plan approved by the Company’s board of directors on the Effective Date. The Kenyon Options have a term of 10 years from the date of grant. 1/3rd of the Kenyon Options shall vest every successive one-year anniversary following the Effective Date, provided, that on each such vesting date Dr. Kenyon is employed by the Company and subject to the other provisions of the Services Agreement. The Initial Kenyon RSUs shall vest on the one-year anniversary of the Effective Date, subject to Dr. Kenyon’s continued employment with the Company through such vesting date. The Additional Kenyon RSUs will vest as follows, subject to Dr. Kenyon’s continued employment with the Company through the applicable vesting date: (i) 4,875 of the Additional Kenyon RSUs shall vest upon the Company submitting the CTA for PRP for the Study in an applicable jurisdiction to be selected by the Company, (ii) 4,875 of the Additional Kenyon RSUs shall vest upon the Company completing an equity financing in the amount of at least $4,000,000 in gross proceeds, (iii) 4,875 of the Additional Kenyon RSUs shall vest upon the shares of the Company’s Common Stock being listed on a senior stock exchange (NYSE, NYSEMKT or NASDAQ), and (iv) the remaining 4,875 of the Additional Kenyon RSUs shall vest upon the Company enrolling its first patient in the Study. Each vested Kenyon RSU shall be settled by delivery to Mr. Kenyon of one share of the Company’s common stock and/or the fair market value of one share of common stock in cash, at the sole discretion of the Company’s board of directors and subject to the Plan, on the first to occur of: (i) the date of a Change of Control (as defined in the Services Agreement), (ii) the date that is ten business days following the vesting of such Kenyon RSU, (iii) the date of Dr. Kenyon’s death or Disability (as defined in the Services Agreement), and (iv) Dr. Kenyon’s employment being terminated either by the Company without Cause or by Dr. Kenyon for Good Reason (as defined in the Services Agreement). In the event of a Change of Control (as defined in the Services Agreement), 50% of any unvested portion of the Kenyon Options and the Kenyon RSUs shall vest immediately prior to such event.
The 39,000 restricted stock units were valued at the fair value of $4.25 per unit or $165,750 based on the quoted trading price on the date of grant. The 19,500 stock options were valued using a Black-Scholes model with the following assumptions: stock price at valuation date of $4.25 based on quoted trading price on date of grant, exercise price of $4.25, dividend yield of zero, years to maturity of 10.00, a risk free rate of 2.42%, and expected volatility 268% for a total value of $82,873 (see Note 8 – Stockholders’ Deficit).
NOTE 11 – CONCENTRATIONS AND RISKS
Concentration of Credit Risk
The Company maintains its cash in banks and financial institutions in Australia. Bank deposits in Australian banks are uninsured. The Company has not experienced any losses in such accounts through June 30, 2020.
The Company currently primarily relies on funding from three convertible debt lenders. Proceeds received in the year from each of the three lenders were $621,000, $362,000 and $200,000, respectively, which represents approximately 45%, 26% and 15%, respectively of total proceeds received by the Company during fiscal year 2019.
The Company currently primarily relies on funding from three convertible debt lenders. Proceeds received in the year from each of the three lenders were $285,000, $505,000, and $227,000, respectively, which represents approximately 18%, 32% and 14%, respectively of total proceeds received by the Company during fiscal year 2020.
Receivable Concentration
As of June 30, 2020 and 2019, the Company’s receivables were 100% related to reimbursements on GST taxes paid.
Patent and Patent Concentration
The Company has filed multiple patent applications relating to its lead product, PRP. The Company’s lead patent application has been granted and remains in force in the United States, Belgium, Czech Republic, Denmark, France, Germany, Ireland, Italy, Netherlands, Portugal, Spain, Sweden, Switzerland, Liechtenstein, Turkey, United Kingdom, Australia, China, Japan, Indonesia, Israel, New Zealand, Singapore, Malaysia, South Africa, Mexico, Republic of Korea and India. In Brazil and Canada, the patent application remains under examination.
In 2016 and early 2017, we filed other patent applications. Three applications were filed under the Patent Cooperation Treaty (the “PCT”). The PCT assists applicants in seeking patent protection by filing one international patent application under the PCT, applicants can simultaneously seek protection for an invention in over 150 countries. Once filed, the application is placed under the control of the national or regional patent offices, as applicable, in what is called the national phase. One of the PCT applications filed in November 2016, entered national phase in July 2018 and another PCT application is currently entering national phase in August 2018. A third PCT application entered the national phase in October 2018.
| F-46 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
Further patent applications are expected to be filed to capture and protect additional patentable subject matter based on the Company’s field of technology relating to pharmaceutical compositions of proenzymes for treating cancer.
Foreign Operations
As of June 30, 2020 and 2019, the Company’s operations are based in Camberwell, Australia, however the majority of research and development is being conducted in the European Union.
On July 22, 2016, the Company formed a wholly owned subsidiary, Propanc (UK) Limited under the laws of England and Wales for the purpose of submitting an orphan drug application with the European Medicines Agency as a small and medium-sized enterprise. As of June 30, 2020 and 2019, there has been no activity within this entity.
NOTE 12 - DERIVATIVE FINANCIAL INSTRUMENTS AND FAIR VALUE MEASUREMENTS
Derivative Financial Instruments:
The Company applies the provisions of ASC 815-40, Contracts in Entity’s Own Equity, under which convertible instruments and warrants, which contain terms that protect holders from declines in the stock price (reset provisions), may not be exempt from derivative accounting treatment. As a result, warrants and embedded conversion options in convertible debt are recorded as a liability and are revalued at fair value at each reporting date. If the fair value of the warrants exceeds the face value of the related debt, the excess is recorded as derivative expense in operations on the issuance date. The Company had $126,500 (3 notes) and $142,000 (2 notes) of convertible debt, which have embedded conversion options bifurcated and is treated as derivative instruments outstanding at June 30, 2020 and 2019 respectively.
The Company calculates the estimated fair values of the liabilities for derivative instruments using the Binomial Trees Method. The closing price of the Company’s common stock at June 30, 2020, the last trading day of the fiscal year ended June 30, 2020, was $0.0039. Volatility, expected remaining term and risk-free interest rates used to estimate the fair value of derivative liabilities at June 30, 2020 are indicated in the table that follows. The expected term is equal to the remaining term of the warrants or convertible instruments and the risk free rate is based upon rates for treasury securities with the same term.
Convertible Debt
| Initial
Valuations | June 30, 2020 | June 30, 2019 | ||||||||||
| Volatility | 227- 279 | % | 264 | % | 355 | % | ||||||
| Expected remaining term | 1.00 | 0.01 – 0.70 | 0.11 - 0.90 | |||||||||
| Risk-free interest rate | 1.53 – 1.59 | % | 0.13 – 0.18 | % | 1.92 - 2.15 | % | ||||||
| Expected dividend yield | None | None | None | |||||||||
Fair Value Measurements:
The Company measures and reports at fair value the liability for derivative instruments. The fair value liabilities for embedded conversion options have been recorded as determined utilizing the Binomial Trees model. The following tables summarize the Company’s financial assets and liabilities measured at fair value on a recurring basis as of June 30, 2020:
Balance at June 30, 2020 | Quoted Prices in Active Markets for Identical Assets | Significant Other Observable Inputs | Significant Unobservable Inputs | |||||||||||||
| (Level 1) | (Level 2) | (Level 3) | ||||||||||||||
| Embedded conversion option liabilities | $ | 177,009 | $ | — | $ | — | $ | 177,009 | ||||||||
| Total | $ | 177,009 | $ | — | $ | — | $ | 177,009 | ||||||||
| F-47 |
PROPANC BIOPHARMA, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2020 and 2019
The following tables summarize the Company’s financial assets and liabilities measured at fair value on a recurring basis as of June 30, 2019:
Balance at June 30, 2019 | Quoted Prices in Active Markets for Identical Assets | Significant Other Observable Inputs | Significant Unobservable Inputs | |||||||||||||
| (Level 1) | (Level 2) | (Level 3) | ||||||||||||||
| Embedded conversion option liabilities | $ | 698,264 | $ | — | $ | — | $ | 698,264 | ||||||||
| Total | $ | 698,264 | $ | — | $ | — | $ | 698,264 | ||||||||
The following is a roll forward for the years ended June 30, 2020 and 2019 of the fair value liability of price adjustable derivative instruments:
| Fair Value of | ||||
| Liability for | ||||
| Derivative | ||||
| Instruments | ||||
| Balance at June 30, 2018 | $ | 371,532 | ||
| Effects of foreign currency exchange rate changes | - | |||
| Reductions due to conversions | (1,388,764 | ) | ||
| Reductions due to repayment of debt | (936,650 | ) | ||
| Initial fair value of embedded conversion option derivative liability recorded as debt discount | 180,000 | |||
| Initial fair value of embedded conversion option derivative liability recorded as change in fair value of embedded conversion option | 382,944 | |||
| Change in fair value included in statements of operations | 2,089,202 | |||
| Balance at June 30, 2019 | 698,264 | |||
| Reductions due to conversions | (362,962 | ) | ||
| Initial fair value of embedded conversion option derivative liability recorded as debt discount | 227,000 | |||
| Initial fair value of embedded conversion option derivative liability recorded as derivative expense | 351,461 | |||
| Change in fair value included in derivative expense (income) in the statement of operations | (736,754 | ) | ||
| Balance at June 30, 2020 | $ | 177,009 | ||
NOTE 13 – SUBSEQUENT EVENTS
Exercise of Warrants
On July 22, 2020, the Company received aggregate gross proceeds of $101,045 from the exercise of 10,445,482 prefunded warrants and 2,500,000 Series B Warrants (see Note 8).
Note Conversions
From July 1, 2020 through September 28, 2020, the Company issued an aggregate of 442,031,352 shares of its common stock at an average contractual conversion price of $0.00096, ranging from $0.00056 to $0.0020, as a result of the conversion of principal of $391,935, interest of $25,735 and conversion fees $6,750 underlying certain outstanding convertible notes converted during such period. On September 18, 2020 the Company prepaid an outstanding convertible note in the amount of $57,671 comprising the note principal of $43,000 plus $1,705 of accrued interest and a prepayment penalty of $12,966.
The Company reclassified $204,919 in put premiums to additional paid in capital following conversions between July 2020 and September 2020.
Notes totaling $75,000 contained bifurcated embedded conversion option derivatives. Accordingly, the fair market value of the shares issued was $134,155 resulting in a loss on extinguishment at the time of conversion of $56,155 and $106,141 of derivative fair value was recorded as a gain on extinguishment at the time of conversion.
| F-48 |
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution
The following table sets forth the costs and expenses, other than underwriting discounts and commissions, to be paid by the Registrant in connection with the issuance and distribution of the securities being registered. All amounts other than the SEC registration fees and FINRA fees are estimates.
| SEC Registration Fee | $ | 2,336.50 | ||
| FINRA Filing Fee | $ | - | ||
| Printing Fees and Expenses* | $ | - | ||
| Accounting Fees and Expenses* | $ | 10,000.00 | ||
| Legal Fees and Expenses* | $ | 15,000.00 | ||
| Transfer Agent and Registrar Fees | $ | - | ||
| Miscellaneous Fees and Expenses* | $ | 1,000 | ||
| Total* | $ | 28,336.50 |
* Estimated expenses.
Item 14. Indemnification of Directors and Officers
Our Certificate of Incorporation contains provisions that limit the liability of our directors for monetary damages to the fullest extent permitted by Delaware law. Consequently, our directors will not be personally liable to us or our stockholders for monetary damages for any breach of fiduciary duties as directors, except liability for:
| ● | any breach of the director’s duty of loyalty to us or our stockholders; | |
| ● | any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; | |
| ● | unlawful payments of dividends or unlawful stock repurchases or redemptions as provided in Section 174 of the Delaware General Corporation Law; or | |
| ● | any transaction from which the director derived an improper personal benefit. |
Our Certificate of Incorporation provides that we are required to indemnify our directors and officers, in each case to the fullest extent permitted by Delaware law. Our Certificate of Incorporation also provides that we are obligated to advance expenses incurred by a director or officer in advance of the final disposition of any action or proceeding, and permit us to secure insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in that capacity regardless of whether we would otherwise be permitted to indemnify him or her under the provisions of Delaware law.
The limitation of liability and indemnification provisions in our Certificate of Incorporation may discourage stockholders from bringing a lawsuit against our directors and officers for breach of their fiduciary duty. They may also reduce the likelihood of derivative litigation against our directors and officers, even though an action, if successful, might benefit us and other stockholders. Further, a stockholder’s investment may be adversely affected to the extent that we pay the costs of settlement and damage awards against directors and officers as required by these indemnification provisions.
In addition, in the future, we intend to enter into indemnification agreements with our directors and officers and some of our executives may have certain indemnification rights arising under their employment agreements with us. These indemnification agreements may require us, among other things, to indemnify our directors and officers for some expenses, including attorneys’ fees, judgments, fines and settlement amounts incurred by a director or officer in any action or proceeding arising out of his or her service as one of our directors or officers, or any of our subsidiaries or any other company or enterprise to which the person provides services at our request.
The above discussion of our Certificate of Incorporation and Delaware law is not intended to be exhaustive and is respectively qualified in its entirety by such bylaws and applicable Delaware law.
These indemnification provisions and the indemnification agreements may be sufficiently broad to permit indemnification of our officers and directors for liabilities, including reimbursement of expenses incurred, arising under the Securities Act. We have been advised that, in the opinion of the SEC, indemnification of directors or officers for liabilities arising under the Securities Act is against public policy and, therefore, such indemnification provisions may be unenforceable.
| 104 |
We maintain a general liability insurance policy that covers certain liabilities of directors and officers of our corporation arising out of claims based on acts or omissions in their capacities as directors or officers.
Item 15. Recent Sales of Unregistered Securities
During the last three completed fiscal years and to date in the current fiscal year, we sold the following unregistered securities:
Shares Issued for Services
On October 27, 2016, we entered into an agreement with a third party for professional services to us over a six-month period commencing on October 10, 2016 in exchange for a monthly fee of $22,500, of which $10,000 a month was to be paid in cash and $12,500 per month in shares of our common stock. Additionally, we acknowledged an existing outstanding balance due of $20,500 for September 2016 services provided by such party to us. On March 2, 2017, we issued 60 shares of our common stock to such third party as consideration for the $75,000 of consulting expenses provided to us.
On February 1, 2017, we received an invoice for $30,000 from a third party for six months of consulting services performed during the period of August 1, 2016 through January 31, 2017. The invoice was payable 50% in cash and 50% in shares of our common stock. We issued 60 shares on July 25, 2017 as the share consideration due under such agreement.
On May 10, 2017, we entered into a seven-month agreement with a third party for growth strategy consulting services to be provided to us, pursuant to which we agreed to issue 15 shares of our common stock per month to such third party as consideration for the services. In June of 2018, the shares were determined by our board of directors to be non-issuable due to non-performance and such shares were not issued under the agreement.
On June 29, 2017, the May 7, 2015 consulting agreement was further amended, and we agreed to issue the consultant 200 shares of our common stock as consideration to eliminate the monthly retainer fee of $7,500 on a going forward basis and to waive the consultant’s right of first refusal to act as lead book running manager of any of our public or private offering of securities or any other financing during the term of the engagement. The 200 shares were issued on July 25, 2017.
On August 1, 2017, we received an invoice for $30,000 from a third party for six months of consulting services provided to us during the period of February 1, 2017 through July 31, 2017. The invoice was payable in shares of our common stock. On February 15, 2018, we issued 469 shares of our common stock to such consultant as payment for the services provided.
On December 29, 2017, we entered into a one-year consulting agreement with a consultant for certain consulting, advisory and media services to be provided to us. As compensation for such services, among other consideration, we agreed to pay the consultant an upfront fee of 1,000 shares of our common stock, with up to 1,500 additional shares to be issued on the six-month anniversary of the date of the consulting agreement at our sole discretion. On February 15, 2018, we issued such 1,000 shares to the consultant.
On February 1, 2018, we received an invoice for $30,000 from a third party for six months of consulting services provided to us during the period of August 1, 2017 through January 31, 2018. The invoice was payable in shares of our common stock. We issued 469 shares of our common stock on February 15, 2018 as payment for such services.
| 105 |
On November 28, 2018, we issued 2,000 shares of our common stock, valued at $30,000, in connection with certain legal services provided to us.
On December 6, 2018, we entered into an agreement with a certain consultant to provide services over a six-month period beginning November 1, 2018 and ending May 1, 2019 in exchange for 4,000 shares of our common stock. On December 27, 2018, we issued such 4,000 shares, valued at $39,000.
On March 5, 2019, we issued the first tranche of 10,000 shares of our common stock to a certain consultant in consideration of certain advisory services provided by the consultant during a three-month period beginning December 11, 2018 and ending March 21, 2019.
On July 19, 2019, the Company entered into an agreement with a certain consultant to provide services over a two-month period beginning July 1, 2019 and ending September 1, 2019 in exchange for 20,000 shares of the Company’s common stock. On July 19, 2019, the Company issued the 20,000 shares of the Company’s common stock valued at $1.99 per share; being the closing price of the stock on the date of the agreement, to such consultant, or $39,800, which will be amortized over the term of the agreement. The Company recorded $39,800 of consulting expense with respect to such shares of its common stock during the three months ended September 30, 2019.
On February 3, 2020, the Company issued 150,000 shares of the Company’s common stock to a consultant for services rendered pursuant to an engagement agreement dated on September 10, 2019 which agreement was later amended in February 2020. On February 3, 2020, the Company issued the 150,000 shares of the Company’s common stock valued at $0.14 per share; being the closing price of the stock on the date of the agreement, to such consultant, or $21,000. The Company recorded $21,000 of consulting expense with respect to such shares of its common stock during the nine months ended March 31, 2020.
Issuance of Shares of Common Stock upon Conversion
Fiscal Year 2017
During our first quarter ended September 30, 2016, we issued 124 shares of our common stock at an average conversion price of $1,240 as a result of the conversion of principal and interest in the aggregate amount of $153,610 underlying certain convertible notes converted during such period.
During our second quarter ended December 31, 2016, we issued 511 shares of our common stock at an average conversion price of $865 as a result of the conversion of principal and interest in the aggregate amount of $424,374 underlying certain convertible notes converted during such period.
| 106 |
During our third quarter ended March 31, 2017, we issued 457 shares of our common stock at an average conversion price of $760 as a result of the conversion of principal and interest in the aggregate amount of $356,257 underlying certain convertible notes converted during such period.
During our fourth quarter ended June 30, 2017, we issued 1,378 shares of our common stock at an average conversion price of $345 as a result of the conversion of principal and interest in the aggregate amount of $472,495 underlying certain convertible notes converted during such period.
Fiscal Year 2018
During our first quarter ended September 30, 2017, we issued 3,536 shares of our common stock at an average conversion price of $135 as a result of the conversion of principal and interest in the aggregate amount of $432,791 underlying certain convertible notes converted during such period.
During our second quarter ended December 31, 2017, we issued 22,664 shares of our common stock at an average conversion price of $45 as a result of the conversion of principal and interest in the aggregate amount of $945,137 underlying certain convertible notes converted during such period.
During our third quarter ended March 31, 2018, we issued 24,845 shares of our common stock at an average conversion price of $35 as a result of the conversion of principal and interest in the aggregate amount of $796,772 underlying certain convertible notes converted during such period.
During our fourth quarter ended June 30, 2018, we issued 30,750 shares of our common stock at an average conversion price of $20 as a result of the conversion of principal and interest in the aggregate amount of $595,488 underlying certain convertible notes converted during such period.
Fiscal Year 2019
During our first quarter ended September 30, 2018, we issued 258,285 shares of our common stock at an average conversion price of $5 as a result of the conversion of principal and interest in the aggregate amount of $1,413,317 underlying certain convertible notes converted during such period.
During our second quarter ended December 31, 2018, we issued 127,684 shares of our common stock at an average conversion price of $10 as a result of the conversion of principal and interest in the aggregate amount of $1,095,100 underlying certain outstanding convertible notes converted during such period.
During our third quarter ended March 31, 2019, we issued 140,673 shares of our common stock at an average conversion price of $4.20, as a result of the conversion of principal and interest in the aggregate amount of $583,331 underlying certain outstanding convertible notes converted during such period.
During our fourth quarter ended June 30, 2019, we issued 177,615 shares of our common stock at an average conversion price of $1.46, as a result of the conversion of principal and interest in the aggregate amount of $259,034 underlying certain outstanding convertible notes converted during such period.
Fiscal Year 2020
During our first quarter ended September 30, 2019, we issued 181,939 shares of our common stock at an average conversion price of $0.68 as a result of the conversion of principal and interest in the aggregate amount of $123,712 underlying certain convertible notes converted during such period.
During our second quarter ended December 31, 2012, we issued 1,064,920 shares of our common stock at an average conversion price of $0.206 as a result of the conversion of principal and interest in the aggregate amount of $219,466 underlying certain outstanding convertible notes converted during such period.
During our third quarter ended March 31, 2020, we issued 16,096,509 shares of our common stock at an average conversion price of $0.053, as a result of the conversion of principal and interest in the aggregate amount of $856,338 underlying certain outstanding convertible notes converted during such period.
During our fourth quarter ended June 30, 2020, we issued 230,275,879 shares of our common stock at an average conversion price of $0.004, as a result of the conversion of principal and interest in the aggregate amount of $925,658 underlying certain outstanding convertible notes converted during such period.
We had 739,138,743 shares of our common stock reserved for future issuances based on lender note conversion requirements pursuant to underlying financing documents at June 30, 2020.
Subsequent Events
Exercise of Warrants
On July 22, 2020, the Company received aggregate gross proceeds of $101,045 from the exercise of 10,445,482 prefunded warrants and 2,500,000 Series B Warrants (see Note 8).
Note Conversions
From July 1, 2020 through September 28, 2020, the Company issued an aggregate of 442,031,352 shares of its common stock at an average contractual conversion price of $0.00096, ranging from $0.00056 to $0.0020, as a result of the conversion of principal of $391,935, interest of $25,735 and conversion fees $6,750 underlying certain outstanding convertible notes converted during such period. On September 18, 2020 the Company prepaid an outstanding convertible note in the amount of $57,671 comprising the note principal of $43,000 plus $1,705 of accrued interest and a prepayment penalty of $12,966.
Other Issuance of Shares of Common Stock
On October 5, 2018, we issued 7,701 shares of our common stock to L2 Capital as the commitment fee under the L2 Equity Purchase Agreement.
On July 19, 2019, we issued 20,000 shares of our common stock to Regal Consulting LLC for services rendered under a Consulting Agreement entered into between the Company and Regal Consulting LLC.
On March 3, 2020 we issued 150,000 shares of our common stock to Sylva International LLC for services rendered under a Consulting Agreement entered into between the Company and Sylva International LLC.
On April 6, 2020 we issued 804,518 shares of our common stock to Ionic Ventures LLC pursuant to a Securities Purchase Agreement.
On April 8, 2020 we issued 1,151,682 shares of our common stock to Sylva International LLC for services rendered under a Consulting Agreement entered into between the Company and Sylva International LLC.
On June 26, 2020 we issued 7,406,892 shares of our common stock to Sylva International LLC for services rendered under a Consulting Agreement entered into between the Company and Sylva International LLC.
| 107 |
Issuance of Restricted Stock Units
On May 14, 2019, our board of directors approved a grant of an aggregate of 78,000 restricted stock units (“RSUs”) to Mr. Nathanielsz, our Chief Executive Officer, Chairman, acting Chief Financial Officer and a director. 39,000 of such RSUs shall vest on the one-year anniversary of the date of the grant, subject to Mr. Nathanielsz being employed by us on such vesting date. The remaining 39,000 RSUs shall vest based on the Company achieving certain milestones as set forth in Mr. Nathanielsz’s Amended and Restated Employment Agreement and summarized elsewhere in this Annual Report. The fair value of the granted RSUs at such grant date was $4.25 per RSU or $331,500 in total based on the closing price of the shares of our common stock on the date of the grant. On May 14, 2019, our board of directors also approved a grant of an aggregate of 39,000 RSUs to Dr. Kenyon, our Chief Scientific Officer and a director. 19,500 of such RSUs shall vest on the one-year anniversary of the date of the grant, subject to Dr. Kenyon being employed by us on such vesting date. The remaining 19,500 RSUs shall vest based on the Company achieving certain milestones as set forth in Dr. Kenyon’s Amended and Restated Services Agreement and summarized elsewhere in this Annual Report. The fair value of the granted RSUs at such grant date was $4.25 per RSU or $165,750 in total based on the closing price of the shares of our common stock on the date of the grant.
Issuance of Options
As of March 31, 2020, the Company had entered into agreements to grant options to purchase 59,644 shares of its common stock, with a weighted average exercise price per share of $76.37.
On April 14, 2016, our board of directors approved a grant of 572 stock options, with an exercise price of $3,750 (market value of the shares of our common stock on such grant date), to each of Mr. Nathanielsz, our Chief Executive Officer, Chief Financial Officer and a director, and Dr. Kenyon, our non-executive director. 191 of such options vested on such grant date and expire on April 14, 2021, 191 of such options vested on April 14, 2017 (first anniversary of such grant date) and expire on April 14, 2021 and 191 of such options vested on April 14, 2018 (second anniversary of such grant date) and expire on April 14, 2021. The fair value of each grant of the 572 options at such grant date was $1,962,440 (aggregate total of $3,924,880).
On May 14, 2019, our board of directors approved a grant of 39,000 stock options to Mr. Nathanielsz, with an exercise price of $4.675 per share, which shall vest annually over a period of three years from the date of the grant, provided, that on each such vesting date Mr. Nathanielsz is employed by us and subject to the other provisions of his Amended and Restated Employment Agreement. On May 14, 2019, our board of directors also approved a grant of 19,500 stock options to Dr. Kenyon, with an exercise price of $4.25 per share, which shall vest annually over a period of three years from the date of the grant, provided, that on each such vesting date Dr. Kenyon is employed by us and subject to the other provisions of his Amended and Restated Services Agreement.
No stock options were issued during the fiscal year ended June 30, 2020.
Issuance of Warrants
In connection with the consulting agreement, dated May 7, 2015, we issued to a consultant 5-year warrants to purchase 27 shares of our common with an exercise price of $3,750 per share.
In connection with the consulting agreement, dated May 21, 2015, we issued to a consultant 5-year warrants to purchase 8 shares of our common stock with an exercise price of $8,750 per share.
On October 28, 2015, pursuant to a convertible debenture, we issued 4-year warrants to purchase 210 shares of our common stock with an exercise price of $75,000 per share.
In connection with the consulting agreement, dated November 11, 2015, on February 22, 2016, we issued to a consultant 5-year warrants to purchase 32 shares of our common stock with an exercise price of $5,625 per share.
On July 8, 2016, the 2015 Warrant to purchase 210 shares of our common stock issued to Delafield Limited Investments (“Delafield”) was fully exercised at a price of $1,500 per share for a total of $314,286 in connection with the Letter Agreement, dated July 1, 2016, entered into with Delafield.
On August 3, 2016, pursuant to the Letter Agreement, dated August 3, 2016 (the “August Letter Agreement”), entered into with Delafield we issued warrants to purchase 1,920 shares of our common stock. 1,600 of these warrants had exercise prices ranging from $1,500 to $2,500 per share and expired five months from the date of issuance. 320 of these warrants had an exercise price of $12,500 per share and expired two years from the date of issuance. These warrants were subsequently cancelled.
| 108 |
On August 18, 2016, pursuant to the August Letter Agreement, warrants to purchase 100 shares of our common stock issued to Delafield were exercised at a price of $1,500 per share under the first tranche of the Five Month Warrant or $150,000 in the aggregate. These shares were subsequently cancelled.
On November 9, 2016, we entered into an agreement (the “November Agreement”) to adjust the exercise price of a warrant, issued on September 30, 2013, to purchase 24 shares of our common stock. Under the terms of the November Agreement, the exercise price for the shares underlying the warrant was reduced to $1,440 per share. The November Agreement did not affect the remaining terms of the warrant.
On December 12, 2016, pursuant to the December Letter Agreement, dated December 2, 2016, entered into with Delafield, we issued a 2-year warrant to purchase 208 shares of our common stock with an exercise price of $6,250 per share.
On August 30, 2019, pursuant to a Securities Purchase Agreement, we issued to Auctus Fund LLC (i) a Common Stock Purchase Warrant permitting the holder to purchase 450,000 shares of common stock of the Company for an exercise price of $2.25, (ii) a Common Stock Purchase Warrant permitting the holder to purchase 300,000 shares of common stock of the Company for an exercise price of $3.33, and (iii) a Common Stock Purchase Warrant permitting the holder to purchase 225,000 shares of common stock of the Company for an exercise price of $4.50.
On September 10, 2019, the Company entered into an agreement with a certain consultant to provide services over a three-month period beginning September 10, 2019 and ending December 10, 2019 in exchange for 1,000,000 warrants to purchase the Company’s common stock at $2.00 per share with an expiry date of September 10, 2022. The Fair Market Value of the warrants was $984,810 on the date of grant as calculated under the Black Scholes Option Pricing model. The Company recorded $984,810 of share based compensation expenses with respect to the grant of such warrants during the six months ended December 31, 2019.
On March 30, 2020, the Company entered into a Securities Purchase Agreement (the “Securities Purchase Agreement”) whereby an investor (the “Investor”) purchased from the Company, 7,500,000 units (the “Units”), each consisting of (i) 1.5 shares of the Company’s common stock (the “Common Stock”), or pre-funded warrants (the “Prefunded Warrants”) and (ii) 1.5 warrants to purchase one share of Common Stock (“Series A Warrants”, and collectively with the Common Stock the “Units”). In addition to the Units, the Investor was issued 63,750,000 warrants to purchase one share of Common Stock (the “Series B Warrants”) and an additional 63,750,000 warrants to purchase one share of Common Stock, subject to a vesting schedule (the “Series C Warrants” and, together with the Prefunded Warrants, the Series A Warrants, and the Series B Warrants, the “Warrants”). The aggregate purchase price for the Units, the Series A Warrants, the Series B Warrants and the Series C Warrants of $450,000 was paid at closing (the “Purchase Price”). The 11,250,000 shares of Common Stock underlying the Units issuable at closing of the Securities Purchase Agreement are comprised of 804,518 shares of restricted Common Stock and 10,445,482 Prefunded Warrants.
As of June 30, 2020, there were 151,170,514 warrants outstanding and exercisable with expiration dates commencing August 2020 and continuing through August 2024, with a weighted average exercise price per share of $0.15.
Except as otherwise noted, the securities in the transactions describe above were sold in reliance on the exemption from registration provided in Section 4(a)(2) of the Securities Act for transactions not involving any public offering. Each of the persons acquiring the foregoing securities was an accredited investor (as defined in Rule 501(a) of Regulation D) and confirmed the foregoing and acknowledged, in writing, that the securities must be acquired and held for investment. All certificates evidencing the shares sold bore a restrictive legend. No underwriter participated in the offer and sale of these securities, and no commission or other remuneration was paid or given directly or indirectly in connection therewith. The proceeds from these sales were used for general corporate purposes.
Item 16. Exhibits and Financial Statement Schedules
(a) Exhibits
We have filed the exhibits listed on the accompanying Exhibit Index of this registration statement and below in this Item 16:
| 109 |
| 110 |
| 111 |
| 112 |
| 113 |
| 114 |
| 115 |
| 116 |
| 117 |
| * | Filed herewith. | |
| ** | To be filed by amendment. |
(b) Financial Statement Schedules.
All schedules have been omitted because either they are not required, are not applicable or the information is otherwise set forth in the financial statements and related notes thereto.
Item 17. Undertakings
The undersigned registrant hereby undertakes:
| (1) | To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement: |
| (i) | To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933; | |
| (ii) | To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and | |
| (iii) | To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement. |
| 118 |
| (2) | That for the purpose of determining any liability under the Securities Act of 1933 each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. | |
| (3) | To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering. | |
| (4) | That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use. |
| (5) | That, for the purpose of determining liability of the registrant under the Securities Act of 1933 to any purchaser in the initial distribution of the securities: | |
| The undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser: |
| (i) | Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424; | |
| (ii) | Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant; | |
| (iii) | The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and | |
| (iv) | Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser. |
| (6) | The undersigned Registrant hereby undertakes to provide to the underwriters at the closing specified in the underwriting agreement certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser. | |
| (7) | Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the Registrant pursuant to the provisions described in Item 14 above, or otherwise, the Registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer or controlling person of the Registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue. | |
| (8) | The undersigned Registrant hereby undertakes: |
| (1) | That for purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4), or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective. | |
| (2) | That for the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and this offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| 119 |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the Registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Melbourne, in the State of Victoria, Australia, on October 13, 2020.
| PROPANC BIOPHARMA, INC. | ||
| By: | /s/ James Nathanielsz | |
| Name: | James Nathanielsz | |
| Title: | Chief Executive Officer (Principal Executive Officer) | |
Pursuant to the requirements of the Securities Act of 1933, this Registration Statement has been signed by the following persons in the capacities and on the dates indicated:
| Signature | Title | Date | ||
| /s/ James Nathanielsz | Chief Executive Officer (Principal Executive | October 13, 2020 | ||
| James Nathanielsz | Officer), Director | |||
| /s/ Carlo Campiciano | Chief Financial Officer (Principal Accounting Officer) | October 13, 2020 | ||
| Carlo Campiciano |
| 120 |