UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended June 30, 2014
Or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ____________ to ______________
Commission file number 001-32521
Propanc Health
Group
Corporation
(Exact name
of registrant as
specified in its charter)
| Delaware | 33-0662986 | |
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
Level 13, Suite 1307, 530 Collins Street
Melbourne, VIC, 3000, Australia
(Address of principal executive offices) (Zip Code)
61 03 9614 2795
(Registrant’s telephone number, including area code)
Securities registered under Section 12(b) of the Act: None
Securities registered under Section 12(g) of the Act:
| Title
of each class registered: |
Name
of each exchange on which registered: | |
| Common Stock, $0.001 par value | Over-the-Counter Bulletin Board |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes oNo x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ¨ | Accelerated filer | ¨ |
| Non-accelerated filer | ¨ | Smaller reporting company | x |
| (Do not check if smaller reporting company) | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No x
The aggregate market value of the Company’s common stock held by non-affiliates computed by reference to the closing bid price of the Company’s common stock, as of the last business day of the registrant’s most recently completed second fiscal quarter: $5,182,094.20.
Indicate the number of shares outstanding of each of the registrant's classes of common stock, as of the latest practicable date: 82,444,100 shares of common stock, par value $0.001 per share, issued and outstanding as of October 14, 2014.
TABLE OF CONTENTS
| 2 |
Forward-Looking Statements
Certain statements in this Annual Report on Form 10-K constitute “forward-looking statements” made under the “safe harbor” provisions of the Private Securities Litigation Reform Act of 1995 that are based on current expectations, estimates, forecasts and assumptions and are subject to risks and uncertainties. Words such as “anticipate,” “assume,” “believe,” “estimate,” “expect,” “goal,” “intend,” “plan,” “project,” “seek,” “target,” and variations of such words and similar expressions are intended to identify such forward-looking statements. All forward-looking statements speak only as of the date on which they are made. Such forward-looking statements are subject to certain risks, uncertainties and assumptions relating to certain factors that could cause actual results to differ materially from those anticipated in such statements.
We cannot predict all of the risks and uncertainties. Accordingly, such information should not be regarded as representations that the results or conditions described in such statements or that our objectives and plans will be achieved and we do not assume any responsibility for the accuracy or completeness of any of these forward-looking statements. These forward-looking statements are found at various places throughout this Annual Report on Form 10-K and include information concerning possible or assumed future results of our operations, including statements about potential acquisition or merger targets; business strategies; future cash flows; financing plans; plans and objectives of management; any other statements regarding future acquisitions, future cash needs, future operations, business plans and future financial results, and any other statements that are not historical facts.
These forward-looking statements represent our intentions, plans, expectations, assumptions and beliefs about future events and are subject to risks, uncertainties and other factors. Many of those factors are outside of our control and could cause actual results to differ materially from the results expressed or implied by those forward-looking statements. In light of these risks, uncertainties and assumptions, the events described in the forward-looking statements might not occur or might occur to a different extent or at a different time than we have described. You are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date of the Annual Report on Form 10-K. All subsequent written and oral forward-looking statements concerning other matters addressed in this Annual Report on Form 10-K and attributable to us or any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this Annual Report on Form 10-K.
Except to the extent required by law, we undertake no obligation to update or revise any forward-looking statements, whether as a result of new information, future events, a change in events, conditions, circumstances or assumptions underlying such statements, or otherwise.
| 3 |
General
As used in this Annual Report, references to “the Company”, “Propanc”, “we”, “our”, “ours” and “us” refer to Propanc Health Group Corporation and consolidated subsidiaries, unless otherwise indicated. In addition, references to our “financial statements” are to our consolidated financial statements except as the context otherwise requires.
We prepare our financial statements in United States dollars and in accordance with generally accepted accounting principles as applied in the United States, referred to as U.S. GAAP. In this Annual Report, references to “$” and “dollars” are to United States dollars.
Overview
We are a development stage healthcare company that is currently focused on developing new cancer treatments for patients, suffering from pancreatic and colorectal cancer. Together with our scientific and oncology consultants, we have developed a rational, composite formulation of anti-cancer compounds, which together exert a number of effects designed to control or prevent tumors recurring and spreading through the body. Our leading products are variations upon our novel formulation and involve or employ pro-enzymes, which are inactive precursors of enzymes. As a result of positive early indications of the anti-cancer effects of our technology, we intend to submit our pro-enzyme treatment to the rigorous, formal non-clinical and clinical development and trial processes required to obtain the regulatory approval necessary to commercialize it and any product(s) derived and/or to be derived therefrom.
In the near term, we intend to target patients with limited remaining therapeutic options for the treatment of solid tumors such as colorectal or pancreatic tumors. In the future, we intend to development our lead product to treat (i) early stage cancer and (ii) pre-cancerous diseases and (iii) as a preventative measure for patients at risk of developing cancer based on genetic screening.
Key Highlights of this opportunity are:
| · | Potential cancer treatment: a once-daily pro-enzyme treatment as a clinically proven therapeutic option in cancer treatment and prevention. |
| · | Multiple mechanisms of action on cancerous or carcinogenic cells: our treatment exerts multiple effects on cancerous cells which inhibits tumor growth and potentially stop the tumor from spreading through the body in contrast to cancer treatments currently available that lack sufficient efficacy to achieve a durable clinical response by preventing tumor recurrence, or inhibiting new growths which spread through the body. As we progress our research, we intend to elucidate further the multiple mechanisms of action to identify opportunities to expand our intellectual property portfolio. Furthermore, we hope to uncover the molecular targets of the pro-enzymes to identify potential opportunities for developing new compounds. |
| · | Encouraging data from patient treatment: Scientific research undertaken over the last 15 years and clinical experience from treating patients in the United Kingdom (the “UK”) and Australia has provided evidence that PRP may be an effective treatment against cancer and warrants further development. |
| 4 |
| · | Unique intellectual property: We are focusing on building a significant portfolio of intellectual property around the use of pro-enzymes in the treatment of cancer, identifying new formulations, alternative routes of administration and potential new therapeutic targets. The PRP drug product is an enhanced pro-enzyme formulation comprising amylase and pro-enzymes of trypsinogen and chymotrypsinogen, in a specific ratio which synergistically enhances the anti-cancer effects of the pro-enzymes compared to when used as singular entities. Patent protection is currently being sought for this PRP drug product, which forms part of the subject matter of International (PCT) Patent Application No. PCT/AU2010/001403 filed on October 22, 2010 in the name of Propanc Pty Ltd. This international PCT application also includes the priority filings of Australian provisional patent application # 2009905147 and # 2010902655, which were filed on October 22, 2009 and June 17, 2010 respectively (as discussed under the section “Intellectual Property”). The PRP-DCM drug product also forms part of the subject matter of International (PCT) Patent Application No. PCT/AU2010/001403. The Authorized Officer indicated in the Written Opinion issued for this international PCT application, that the patent claims covering the PRP and PRP-DCM products were novel over the prior art cited in the International Search Report. Various national phase applications are being filed in countries around the world based on the above priority applications. |
| · | Market opportunity: Growing demand for new cancer treatments as a result of a rapidly aging population and changing environmental factors in western countries. According to the World Health Organization, all cancers (excluding non-melanoma skin cancer) are expected to increase from 8.2 million annual deaths in 2012 to over 10 million annual deaths by 2020, exceeding 13 million annual deaths by 2030. |
Company History
Propanc Health Group Corporation, formerly Propanc PTY Ltd., is a development stage enterprise and was incorporated in Melbourne, Victoria Australia on October 15, 2007. Based in Melbourne, Victoria Australia, since inception, substantially all of the efforts of our company have been the development of new cancer treatments targeting high risk patients who need a long term therapy which prevents the cancer from returning and spreading. The Company anticipates establishing global markets for its technologies.
On November 23, 2010, Propanc Health Group Corporation was incorporated in the state of Delaware. In January 2011, to reorganize the Company, Propanc Health Group Corporation acquired all of the outstanding shares of Propanc PTY Ltd. on a one-for-one basis making it a wholly-owned subsidiary.
We were formed for the specific purpose of having shareholders of Propanc PTY Ltd. directly owning an interest in a U.S. company. On January 29, 2011, we issued 64,700,525 shares of our common stock in exchange for 64,700,525 shares of Propanc PTY Ltd. common stock.
Company History
Propanc’s scientific roots date back almost 100 years to the work of Professor John Beard at the University of Edinburgh in the United Kingdom (the “UK”) whose pioneering work on tumor cell biology and potential new approaches to treating cancer by targeting specific pathways which kill off cancer cells, but leave healthy cells alone. In more recent times interest in the work of Professor Beard has re-emerged, driven by the insights into his work offered with modern day knowledge of tumor cell and molecular biology.
| 5 |
Important Milestones for Propanc
| · | From the late 90’s, work from other scientists and clinicians, including Dr. Josef Novak in the U.S. and a since retired oncologist, Dr. Frantisek Trnka, from the Czech Republic, shed new light on the therapeutic potential of Professor Beard’s insights. Extensive laboratory work undertaken over a number of years by Novak and Trnka was reported in the journal Anticancer Research in 2005 in the paper entitled ‘Pro-enzyme Therapy of Cancer’. The conclusion of Novak and Trnka from this work was the discovery “that pro-enzyme therapy mandated first by John Beard nearly one hundred years ago, shows remarkable selective effects that result in growth inhibition of tumor cells with metastatic potential”. Today, these important scientific observations support our view that pro-enzymes are selective and effective in targeting malignant tumor cells and could become an effective tool in the fight against metastatic cancer. |
| · | In 2007, Dr. Julian Kenyon, Medical Director of the Dove Clinic in the United Kingdom and Dr. Douglas Mitchell, further developed the therapeutic concepts of Beard and identified strategies which could improve upon the therapeutic potential of Beard’s original ground-breaking work. A suppository formulation was developed by Mandeville Medicines, Buckinghamshire, UK, at the request of, and in consultation with, Dr. Kenyon and Dr. Mitchell, in an effort to improve on results reported in the literature pertaining to the potential therapeutic use of pro-enzymes in cancer treatment. Patients were first treated with the suppository formulation in April 2007 at The Dove Clinic, UK and in July 2007 at the Opal Clinic, Australia. Drs. Kenyon and Mitchell, through The Dove Clinic and Opal Clinic respectively, treated cancer patients in the United Kingdom and Australia with a suppository formulation of pro-enzymes. The treatment was undertaken under special UK and Australian regulatory provisions. In the UK it was undertaken under the Medicines and Healthcare Products Regulatory Agency (the “MHRA”)’s regulations designed for patients who have special clinical needs that cannot be met by licensed medicinal products, and in Australia under the Therapeutic Goods Administration or TGA Special Access Scheme, a mechanism which provides for the import and/or supply of an unapproved therapeutic good for a single patient, on a case by case basis. In both jurisdictions, patients are permitted to receive treatment on an individual basis for compassionate use as long it is supplied by a recognized, licensed manufacturer who is able to meet certain guidelines for unapproved products, and individual case files are maintained for patients should the regulatory authorities require this information. No prior approval was required by either the MHRA or TGA prior to the commencement of treatment. No suppository formulation of the pro-enzymes was available and it was necessary for a novel suppository formulation to be manufactured specifically for these patients by a suitably licensed manufacturer. |
| · | Forty-six late stage cancer patients suffering from a range of malignancies in the UK and Australia received treatment with the pro-enzyme suppositories over periods of time ranging from one (1) month to in excess of seventeen (17) months. Inspired by their observations in clinical practice, Dr. Kenyon and Dr. Mitchell resolved to develop pro-enzyme therapy for cancer patients worldwide. |
| · | Late 2007, Dr. Kenyon, Dr. Mitchell and Mr. James Nathanielsz, our Chief Executive Officer, developed a strategy to commercialize the newly developed pro-enzyme formulation, now designated PRP. Propanc Pty Ltd, a subsidiary of Propanc Health Group Corporation, was established in Australia to refine, develop and commercialize novel, patented pro-enzyme therapeutics for the treatment of cancer. This remains our intention to date. |
| · | In 2008, a Scientific Advisory Board (the “Advisory Board”) comprising Professor John Smyth (Edinburgh University), Professor Klaus Kutz (Bonn University) and Professor Karrar Khan (De Montfort University) was established. Dr Ralf Brandt, Chief Executive Officer and Founder of preclinical Contract Research Organization (CRO), vivoPharm Pty Ltd., was later appointed to the Board in 2011. Today, the expertise of the Advisory Board in oncology research and development will be relied upon as we initiate patient trials and advance our products down the requisite regulatory pathways to commercialize our pro-enzyme therapies. |
| · | In 2009, a retrospective review of the patient notes from the forty-six (46) patients treated was undertaken by Dr Kenyon. This report was subject to analysis by Professor Klaus Kutz who, at the time of the review, was an independent consultant in clinical pharmacology and safety, specializing in oncology. Professor Kutz observed that no patients were reported as living for a period less than that predicted by the treating clinician and a number of terminally ill patients lived marginally longer than predicted, particularly those suffering from pancreatic, colorectal, ovarian and gastro-intestinal cancers. As a result of the observations made by Dr Kenyon and Professor Kutz, we are targeting the development of pro-enzyme therapy for the treatment of colorectal and pancreatic cancers for clinical trials, and in the future targeting other cancer types as our product candidate progresses to commercialization. |
| 6 |
| · | In early 2008, a research collaborative partnership was established with Professor David Tosh, at the Center for Regenerative Medicine, Deparment of Biology and Biochemistry, Bath University, to investigate the molecular mechanisms by which the pro-enzyme formulation is acting, which resulted in us filing two provisional patents a year later. We undertook additional scientific research with Professor Tosh, Dr. Macarena Pèran, Department of Health Sciences, Jaén University, and Dr. Juan Antonio Marchal, Biopathology and Regenerative Medicine Institute, Granada University. Important anti-cancer effects of the pro-enzymes were discovered, including triggering cell necrosis (cell death) and apoptosis (programmed cell death) and significantly, the induction of cell differentiation (i.e. inducing cancer cells to exhibit normal cell behavior). This led to us increasing our intellectual property base and patent new pharmaceutical compositions designed to enhance the effects of pro-enzymes. Subsequently, two provisional patents were combined into one Patent Cooperation Treaty (PCT) Application, filed on 22 October 2010 (PCT Application), and then a year later, we completed a 30 month national phase filing deadline for an international patent and commenced entering the national phase in countries around the world. So far, we received grant status in South Africa and more recently in New Zealand. In addition, the United States Patent and Trademark Office or USPTO and European Patent Office or EPO have made preliminary indications that key features of our technology are patentable. We are presently working towards securing a patent in each region, covering as many aspects of its technology as possible, whilst also actively seeking protection throughout Eastern Europe, Asia and South America. |
| · | Late 2010, we made additional important discoveries and scientific observations, resulting in additional composition claims which were included in the PCT Application, further protecting the company’s pro-enzyme formulation. Collaboration with vivoPharm Pty Ltd. (vivoPharm), located in Melbourne, Australia, with research facilities in Hershey, Pennsylvania, United States, identified a highly synergistic ratio of the pro-enzymes when combined together, resulting in increased anti-cancer effects in several tumor cell lines. By 2011, further work completed by vivoPharm confirmed the anti-metastatic effects of the newly combined ratio of the pro-enzymes in various cell line assays, and anti-angiogenic (inhibition of blood vessel formation) properties of the pro-enzyme treatment in mice. |
| · | In mid-2012, we began trading on the Over the Counter Bulletin Board (“OTCBB”). At the time, whilst located in Melbourne, Australia, we decided to access the US capital markets for raising the capital needed to finance the company’s pro-enzyme treatment for future clinical trials. Today, after deepening our scientific knowledge of the anti-cancer effects of pro-enzymes through our ongoing efforts with our research partners and strengthening our intellectual property portfolio by filing our patents in countries around the world, we are ready to complete the formal animal studies necessary to undertake human trials in 2015. |
| · | In May 2013, it was observed that pro-enzymes enforce the re-entry of cancer cells back into normal cellular pathways and this may represent a novel approach to the treatment of cancer. These findings were published in Cellular Oncology, a peer reviewed journal of the International Society for Cellular Oncology. |
The Problem
In the early phases of tumor progression, cancer cells multiply near the site where their predecessors first began uncontrolled proliferation. The result, usually over a long period of time, is a primary tumor mass. Tumors often need to reach a large size before they make themselves apparent to the individual concerned, or the clinician screening for them.
| 7 |
Eventually, tumors of substantial size may begin to compromise the functioning of organs in which they have arisen and begin to evoke symptoms. In many cases, the effects on normal tissue function come from the physical pressure exerted by the expanding tumor masses. For example, large tumors in the colon may obstruct digestion products through the lumen, or in the lungs, airways may be compromised.
As dangerous and threatening as these primary tumors are, they are ultimately responsible for only about 10% of deaths. A far greater threat often arises for the patient, even after a primary tumor has been identified and removed. This threat involves cancerous growths that are discovered at sites far removed from the locations in their bodies where their primary tumors first appeared. These cancerous growths, called metastases, are responsible for 90% of patient deaths from cancer. Metastases are formed by cancer cells that have left the primary tumor mass and traveled by the body’s blood and lymphatic vessels (a vein like vessel carrying lymph, or white blood cells, from the tissues) to seek new sites and form new colonies. For example, breast cancers often spawn metastatic colonies in many tissues throughout the body including the brain, liver, bones, and lungs.
For primary tumors which have not yet metastasized, current treatments for cancer can be effective in initially reducing tumor burden. However, for many forms of cancer, current treatments lack sufficient efficacy to achieve a long lasting clinical response. Therefore, a vast majority of patients who succumb to cancer are killed by tumors that have metastasized. Continuing with the example of breast cancer, according to the National Cancer Institute’s SEER Cancer Statistics Review (2001 – 2007), of the patients diagnosed with late stage metastatic breast cancer, only 23% are expected to live longer than five years. This is compared to a 98% five year survival rate for an early stage breast cancer patient when the cancer is confined to the primary site.
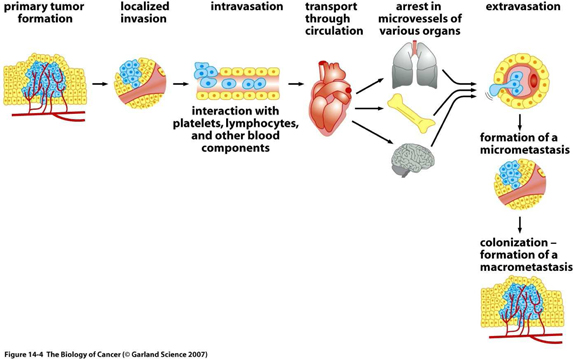
The invasion-metastasis cascade
The great majority of life threatening cancers occur in epithelial tissues, yielding carcinomas. Epithelial cells generally have a multi-sided, uniform shape. They have well defined contact points with neighboring cells and a strong attachment to the underlying connective tissue, or stroma, which creates a framework for solid tumors in the body. Separating the two is the specialized type of extracellular matrix, known as the basement membrane.
By definition, carcinomas which originate on the epithelial side of the basement membrane and are considered to be benign, as long as the cells forming them remain on the same side. However, many carcinomas acquire the ability to penetrate the basement membrane, and individual cancer cells or groups of cancer cells begin to invade the stroma. This mass of cells is now reclassified as malignant. Often, many pathologists and surgeons reserve the label “cancer” for those epithelial tumors that have acquired this invasive ability.
Thereafter, carcinoma cells may invade into lymphatic or blood microvessels. The latter may then transport these cancer cells to distant sites in the body where they may be trapped and subsequently form new metastases.
| 8 |
It is important to note, that even before cells penetrate the basement membrane, they often stimulate angiogenesis (blood vessel formation) on the stromal side of the membrane, by expressing angiogenic proteins through the porous barrier. Not only does this enhance the ability of malignant cells to circulate into the blood, but also provides an important feedback loop for the cancer cell to maintain its invasiveness.
Understanding the mechanism by which benign cells change to a malignant state is therefore pivotal to developing anti-cancer treatments that have sufficient efficacy to achieve a long lasting clinical response.
The epithelial-mesenchymal transition and associated loss of E-cadherin expression enable carcinoma cells to become invasive.
Epithelial cells can undergo a transformation to a different cell type, called mesenchymal cells, through a process called the epithelial-to-mesenchymal transition, or EMT. Mesenchymal cells have an elongated spindle shape; lack orderly contacts with neighboring cells and can survive without contact with a surface or connective tissue. The EMT process is a series of events that normally occur during the development of tissues and organs prior to birth, and also apply to normal wound healing processes. However, the same EMT process can also be applied to epithelial cancer cells, or carcinomas. When epithelial carcinoma cells residing in a solid tumor undergo the EMT process, the resulting mesenchymal cancer cells can invade through local barriers and metastasize to other parts of the body.
In addition to becoming invasive and motile after undergoing the EMT process, the resulting mesenchymal cells have significantly increased resistance to current cancer treatments. For example, in Cancer Research in 2005, it was reported that lung cancer cells expressing mesenchymal biomarkers appeared to be resistant to Tarceva and other targeted anti-cancer agents when transplanted into mice.
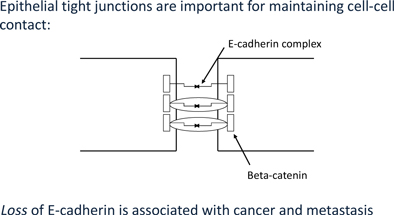
At the center of this critical process for transforming benign cells into carcinomas, is the protein Epithelial Cadherin, or E-Cadherin. In normal cells, E-cadherin is located in the membrane and involved in maintaining cell to cell contact, which is critical to normal function and structure of epithelial tissues. The individual E-Cadherin molecules are attached to the actin (scaffolding, or cytoskeleton structure) within the cell, anchored by β-catenin, a protein which helps form the junction between epithelial cells. As well as forming an anchor between epithelial cells, β-catenin is also involved in gene transcription, a process by which DNA (deoxyribose nucleic acid) is converted into RNA (ribose nucleic acid) within the nucleus of a cell for the purpose of producing new proteins normally associated with routine cell function.
| 9 |
In the case of tumors, when cells become invasive, E-Cadherin expression decreases substantially, β-catenin becomes free within the cell, which may then migrate to the nucleus and induce expression of the EMT program. Furthermore, once cells undergo an EMT, they begin to produce their own cytokines (cell signaling molecules), such as Transforming Growth Factor β, or TGF-β. This protein plays a critical multi-functional role in promoting angiogenesis, immunosuppression (suppressing the immune system from recognizing and attacking cancer cells), and maintaining their mesenchymal cell structure for prolonged periods via a feedback mechanism. Studies also suggest that TGF-β works with β-catenin to cause epithelial cancer cells to undergo an EMT.
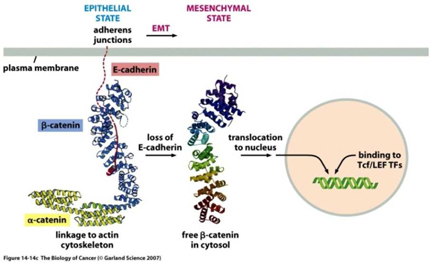
A study in the British Journal of Cancer, 2011, demonstrated that in cholangiocarcinoma (bile duct cancer) cell lines, treatment of TGF-β increased cell migration, invasion and mesenchymal changes. Furthermore, expression of E-cadherin and N-cadherin was measured from resected (cut out) specimens from extra-hepatic (outside the liver) cholangiocarcinoma patients. Patients with low E-cadherin expression had a significantly lower survival rate than patients with high E-cadherin expression. They concluded the cadherin switch via TGF-β induced EMT in extra-hepatic cholangiocarcinoma leads to cancer progression.
| 10 |
Conversely, in studies of several types of carcinoma cells that had lost E-cadherin expression, re-expression of this protein strongly suppressed the invasiveness and motility of these cancer cells.
Together, these observations indicate that E-Cadherin levels is a key determinant of the biological behavior of epithelial cancer cells and that the cell to cell contact constructed by E-cadherin molecules impede invasiveness and hence metastasis.
Our Solution
Our solution is to develop and commercialize a long-term therapy to prevent tumor recurrence and metastases, the main cause of patient death from cancer. We believe this problem can be addressed by developing a pro-enzyme formulation specifically targeting malignant carcinoma cells to and create a long lasting clinical benefit to the patient.
Propanc’s Theory Pro-enzymes Regulate Cell Proliferation
More than 100 years ago, Professor Beard, a comparative embryologist, made an observation that the pancreas develops in most vertebrates at the time when the placenta begins to slow its rate of growth. He hypothesized that enzymes produced by the developing pancreatic gland curtail trophoblastic invasion (A rare condition in which abnormal cells grow inside the uterus from tissue that forms after conception) and suggested that pancreatic extracts should have a similar inhibitory effect on invasive tumors.
Subsequently in the late 90’s, after following Professor Beard’s recommendations, Novak and Trnka hypothesized that administration of pro-enzymes, rather than the enzymes, was of crucial importance to the clinical effectiveness of the treatment approach first developed by Professor Beard, and that the precursor nature of the active enzymes may offer protection against numerous serpins (proteins which can inhibit pro-enzymes) in the blood.
As knowledge of tumor cell and molecular cell biology has increased over the years, our scientists and research partners have made important scientific discoveries identifying that pro-enzymes suppress the EMT program and induce cell differentiation, i.e., return cancerous cells towards normal cell behaviour, or a benign state.
After more than 100 years, the initial observations made by Professor Beard may have a potential common link between embryogenesis and cancer, by which cells are able to become motile and invasive, via the EMT program, where the administration of pro-enzymes may regulate cell proliferation as a means to controlling carcinomas.
Our Product Candidates
We are using our intellectual property and expertise to develop a pro-enzyme therapy for the treatment and prevention of the development of carcinomas from solid tumors. Initially, our products will be used in the treatment of pancreatic and colorectal cancers. In the future, we intend to expand our products scope in anti-cancer treatment to include other common solid tumors such as ovarian, gastrointestinal and prostate cancers.
PRP
Our lead product, PRP, is a novel, patented, formulation consisting of two pro-enzymes; trypsinogen and chymotrypsinogen, plus the enzyme amylase (1, 4-α-D-glucan glucanohydrolase). In limited human testing as described earlier, supplemented by laboratory research at the Universities of Bath and Granada on the mechanism of action of the pro-enzyme mixture, evidence has been obtained which suggests PRP may be effective against a range of solid tumors.
| 11 |
Selectivity
Research published in 2005, suggests that the pro-enzymes in our product, typsinogen and chymotrypsinogen exhibit specificity for tumor cells and not normal cells.. Once activated, they in turn activate Protease Activated Receptors Type 2 (PAR2), which are located on the cell membrane and involved with cancer cell proliferation. Activation of PAR 2 results in a cascade of intracellular activities, including activation of a major component of the cell which controls its structure and architecture, the actin cytoskeleton. In a cancer cell, pro-enzymes have the effect of converting globular actin into filamentous actin, which causes the cell structure to collapse and induce cell death. This reduces tumor volume and is often seen in clinical practice.
In addition, the enzyme amylase contributes to the anti-tumor activity by splitting the carbohydrate element of glycoproteins on the surface of the tumor cell; this action is facilitated by the activated proteases around the cell.
Anti-Cancer Effects and Mechanism of Action
PRP consists of pro-enzymes which are known to influence a number of pathways critical for cancer cells to invade, grow and metastasize. Research published in 2013, shows the clinical benefits of PRP appear to result from enhanced differentiation of tumor cells, which inhibits proliferation and consequently, reduces their ability to invade and metastasize.
Specifically, we showed that pro-enzymes:
| · | induce a dose-dependent inhibition of cell growth, triggering apoptosis and cell necrosis; |
| · | enhance expression of epithelial markers, such as E-cadherin and β catenin; |
| · | decrease expression of EMT transcription factors responsible for coding specific gene sequences from DNA, associated with TGF-β cell signalling pathways; and |
| · | induce malignant cells to differentiate to benign forms. |
Once activated, pro-enzymes influence the micro-immune environment around the cell, altering a number of pathways critical for supporting cancer cell growth, invasion and metastasis. This includes interacting with proteinases and cell signaling pathways in the extracellular matrix, whilst also interacting directly with cell surface proteins that effect the internal pathways of the cancer cell, triggering re-expression of epithelial markers, reducing important EMT markers, and inducing a series of cellular activities which alters the cancer cell’s morphology (structure) from a malignant to a benign state.
Preclinical Development
PRP activates E-Cadherin and β-Catenin Expression, Inhibiting Cell Growth in a Dose Dependent Manner
Initial experiments were performed to determine the effects of PRP on cell growth. Increasing doses of the pro-enzymes in PRP, trypsinogen and chymotrypsinogen were administered at increasing concentrations on three cancer-derived cell lines, including colorectal (Caco-2), pancreatic (Panc1) and esophageal (OE33) carcinomas.
Overall the cell numbers of these three cell lines slightly decreased at concentrations of ≤22 × 103 ng per mL for both trypsinogen and chymotrypsinogen, and ≤5.15 × 103 ng per mL for amylase. However, at ≤28 × 103 ng per mL for both trypsinogen and chymotrypsinogen, and ≤6.15 × 103 ng per mL for amylase, the cell numbers dropped sharply to below 60% and significantly decreased further at higher concentrations, especially for Caco-2 and OE33 carcinoma cell lines. These results suggest that PRP affects cellular growth in a dose dependent manner.
| 12 |
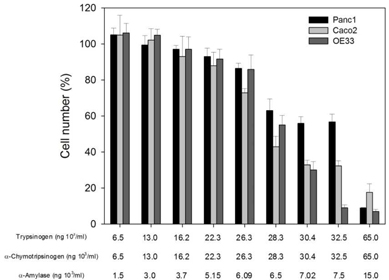
PRP increases the Expression of Epithelial Markers in Carcinomas
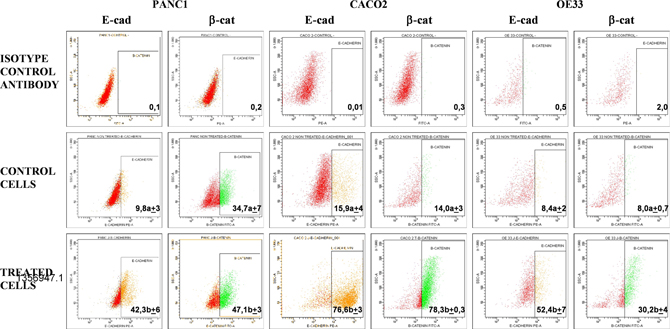
Upon treatment with PRP, changes in expression of the epithelial markers β-catenin and E-cadherin were assessed in Caco-2, Panc1 and OE33 cells. Subsequent flow cytometry analysis (flow cytometry is a laser-based technology that quantitate properties of single cells, one cell at a time) revealed that expression of E-cadherin in Caco-2 cells increased to 76.6% ± 3.0 when cells were treated with PRP, as compared to a control using untreated cells (15.9 % ± 4.2).
Changes in the expression of β-catenin in Caco-2 cells were also observed with an increase from 14.0 % ± 3.5 in control cells to 78.3% ± 0.3 after PRP treatment. E-cadherin expression increased to 42.3 %±6.1 and β-catenin to 47.1% ± 3.3 when Panc1 cells were treated with PRP, while in control cells the respective expression levels were 9.8 %± 2.9 and 34.7 %±7.4. Finally, PRP treated OE33 cells also showed an increment of both epithelial markers compared to untreated control cells, i.e., E-cadherin increased up to 52.4 %±6.8, whereas control cell expression was 8.4 %± 2.1, and β-catenin increased up to 30.2 %±4.2, whereas untreated control cells showed 8.0 %±0.7 expression. In all cases differences between untreated and PRP treated cells were statistically significant (p<0.05).
| 13 |
PRP has proven anti-tumor efficacy in Melanoma (B16-F10) Tumor Bearing Mice
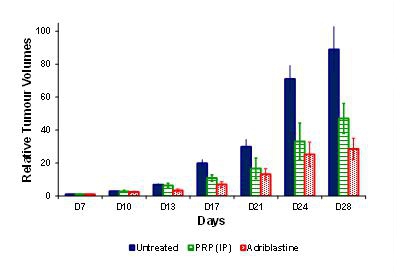
The anti-tumor activity of PRP was assessed in a B16-F10 melanoma model. The tumors were grafted under the skin in C57BL/6 female mice. The tumor-bearing mice were dosed with PRP twice daily, vehicle or control (doxorubicin (Adriblastine™) dosed once at 12 mg/kg i.v (i.e. approximately four-times the clinical dosage based on mg/kg conversion to a 60 kg human) for 28 consecutive days (n = 10 for each group). Treatment began 7 days post-implant.
During the course of the experiment, animals were sacrificed if any of the following occurred: signs of suffering (cachexia, weakening, difficulty moving or eating), compound toxicity (hunching, convulsions, diarrhea), tumor growing to 10% of body weight, tumor ulcerating and remaining open, position of tumor interfering with movement/feeding, 15% body weight loss for 3 consecutive days or 20% body weight loss for 1 day. The mice were sacrificed when the tumor volume reached a maximum volume of 2,000 mm.
Treatment with PRP was well tolerated following intra-peritoneal (i.p) injection into mice. Observations were specifically made to observe any drug-related toxicity (including hunching, convulsions, and diarrhea). There were no adverse events attributable to PRP, nor injection site reactions. Following 21 days treatment (28 days post-implantation), relative tumor volumes (defined as tumor volumes measured x number of days post treatment, divided by the tumor volume measured at day 0, post treatment) were significantly smaller in the i.p. and adriblastine groups compared to the untreated control.
This experiment shows our product, PRP has anti-tumour efficacy in mice, but without the severe, or even serious side effects normally associated with current treatment standards such as chemotherapy.
PRP-DCM
To date, we have been focused on developing a novel combination of anti-cancer agents working in combination with pro-enzymes which enhance PRP’s anti-cancer effects. The enhanced pro-enzymes-based formulations combine PRP with at least one of two types of identified compounds considered on the basis PRP’s mechanism of action to synergistically enhance the anti-cancer effects of PRP.
| 14 |
Our recent work has focused on maximizing the potential of PRP as a drug suitable for long-term maintenance by enhancing the effects of our current pro-enzyme formulation by screening additional active ingredients to enhance the anti-cancer activity of PRP.
Propanc’s scientists believe the additional ingredients identified in the course of this research to augment anti-cancer activity of PRP may also be suited as a stand-alone, adjunct therapy for standard treatment approaches, such as chemotherapy.
Anti-Cancer effects and mechanism of action
Cells obtain the energy they require from aerobic or anaerobic respiration (with, or without oxygen, respectively). It has been suggested that tumor cells rely on anaerobic respiration due to impairment of the mitochondria (an organelle found in most cells, in which the biochemical processes of respiration and energy production occur). We have identified compounds which have pronounced effects on the anaerobic cells within a tumor, which would complement PRP and standard treatment approaches:
| · | 2-deoxy-D-glucose, a metabolite which inhibits glucose metabolism in cancer cells, as reported in the British Journal of Cancer, 2002; |
| · | Capsiate, a non-pungent component from sweet peppers, induces apoptosis by increasing the production of oxygen in cancer cells through forced up-regulation of cell mitochondria, published in the European Journal for Nutrition, 2003; |
| · | Methyl-seleno-cysteine, which at low doses increases the oxidative stress on cancer cells by inhibiting a specific enzyme known to be up-regulated in tumor cells, published in Biochemical Pharmacology, 2008. |
Preclinical Development
In November 2010, we established collaborative research partnership with Dr. Paul Clayton, an expert in cancer prevention and nutrition and former advisor to the Committee on Safety of Medicines (UK), identifying specific anti-cancer agents in combination with one another, and with PRP, enhancing their ability to target cancerous cells with minimal side effects to healthy cells.
As a result of the work undertaken in collaboration with Dr. Paul Clayton, an international PCT application was filed late 2010, detailing enhanced pro-enzyme patent formulations and combination therapies comprising trypsinogen and chymotrypsinogen. Dr. Clayton was awarded a success fee in the form of shares of our common stock representing 1% of the shares then currently issued and outstanding in recognition of his contribution to this research. The patent application is jointly owned by us and the University of Bath, with an exclusive right and license to commercialize any joint intellectual property being held by Propanc (see under License Agreements and Intellectual Property for further details).
Effects on Cell Growth Inhibition Alone and In Combination
The interaction that occurs between agents can be described as synergistic, additive or antagonistic. The work we have conducted to enhance the anti-cancer effects of PRP focused on the positive therapeutic outcome of drug interactions, specifically synergism. The major benefits of additive and synergistic drug interactions are increased efficacy and significantly diminished toxic side effects. This can be achieved by reducing the dose of a drug that elicits damaging side effects, through a combination with another drug. Alternatively, a drug with insufficient efficacy could produce super-additive (synergistic) effects in a well-designed combination.
IC50 determination assays (the concentration of drug to cause 50% reduction in proliferation of cancer cells) were performed for 2-deoxyglucose, capsiate, methyl-seleno-cysteine and the mixture of these three components (i.e. DCM) in a human colorectal carcinoma cell line, HCT-15. IC50 values were obtained for 2-Deoxyglucose, capsiate and DCM. Methyl-seleno-cysteine treatment of the cells resulted in a maximum growth inhibition of 14.8% at the maximum tested concentration and therefore, an IC50 value was not obtained for this Test Article.
| 15 |
Following the IC50 determination assays, a scientific method was employed to study the interaction between 2-deoxy glucose, capsiate and methyl-seleno-cysteine. We found:
| · | capsiate, 2-deoxyglucose and DCM are inhibitors of the growth of the human colorectal carcinoma cell line HCT-15 in vitro; and |
| · | methylselenocysteine and 2-Deoxyglucose synergise to inhibit the growth of the human colorectal carcinoma cell line HCT-15. |
We have also made several other similar observations with other compounds that act to inhibit the growth of the human colorectal carcinoma cell line HCT-15. Further work is needed to assess the optimal combination of ingredients before undertaking formal preclinical development of a potential new combination therapy in animals. We will determine a final combination to be developed as an adjunct therapy to PRP .
Preclinical development
The anti-angiogenic efficacy of PRP in combination with DCM was investigated using vivoPharm's AngioChamber™ assay. The AngioChamber™ assay utilises the normal physiological process of wound healing, to promote fibrous capsule formation around an implanted chamber in mice. The inclusion of Basic Fibroblast Growth Factor (bFGF) in the chamber supports the fibrouse capsule formation while inducing blood vessel development. Thus, this system is used to assess the efficacy of anti-angiogenic treatments by measuring fibrous capsule formation (wet weight of capsule at termination).
Fifty female FvB mice each received a subcutaneously implanted AngioChamber™, with or without bFGF. Ten mice were randomly selected and implanted with Chambers without bFGF. Forty mice which were implanted with Chambers containing bFGF were randomised by body weight into four treatment groups of 10 mice on Day 0 of the study. A reference compound, Sorafenib (60 mg/kg, per oral) was also introduced into the study.
In this Study both treatments resulted in significant inhibition of bFGF-induced angiogenesis compared with the Induction Control, as indicated by the capsule wet weights on the termination day of the study. Both the reference compound, Sorafenib, and the combination of PRP-DCM significantly reduced angiogenesis.
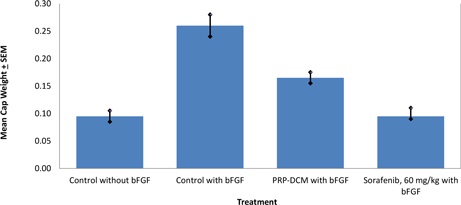
As is frequently seen in cancer research, animal cancer models using PRP and DCM in combination have in some instances shown very encouraging results, with less clear cut results in other animal models. We are working to understand which models are most appropriate, and how to further optimize the DCM formulation as a possible adjunct therapy for use, either in combination with PRP, or other standard treatment approaches.
| 16 |
The PRP Formulation
Oral pancreatic enzymes have been administered previously in a variety of circumstances, and are in current clinical use in conditions where the pancreas is unable to produce sufficient enzymes for the digestion of food. A number of oral pancreatic enzyme products are presently approved in the U.S for use in patients who do not produce enough pancreatic enzymes. Approved pancreatic enzyme products include Pancreaze™ from Johnson & Johnson, CREON® from Abbott Laboratories, and ULTRASE® from Axcan Pharma US.
PRP is a combination of two pro-enzymes trypsinogen and chymotrypsinogen, specifically formulated within a specific ratio designed to synergistically enhance their anti-cancer effects and in combination with other therapies identified based on the mechanism of action. Patent protection is currently being sought for PRP and other potential combinations, which forms part of the subject matter of International (PCT) Patent Application No. PCT/AU2010/001403 filed on 22 October 2010 in the name of Propanc Pty Ltd, our Australian operating subsidiary.
Oral enzymes have also been investigated previously for the treatment of cancer and, whilst generating encouraging results, their widespread use has been hampered by the very large quantities that have been considered necessary for effective treatment – 130 or more tablets per day. The high dose used with oral delivery is considered necessary due to the oral enzymes being broken down in the stomach and duodenum, the first part of the small intestine, and very little actually being absorbed into the general circulation. By administering a pro-enzyme by intravenous or I.V. injection, and using a specific pro-enzyme formulation, the normal breakdown of the enzymes when taken orally is avoided and the drug can potentially be absorbed into the general circulation intact. It is also suggested that pro-enzymes are resistant to inactivation by numerous protein digesting enzymes, like serpins, which are circulating in the blood. Together with our scientific consultants, we believe that the development of an I.V injection pro-enzyme formulation will lead to improved efficacy in the treatment of cancer compared with current oral enzyme preparations, and will substantially reduce the dose in comparison to that used previously for oral enzyme therapy for the treatment of cancer.
Our Research Programs
POP1
In order to maximize our proprietary knowledge on the use of pro-enzymes in the treatment of cancer, we are currently undertaking research to identify the mechanism at the molecular level by which PRP is acting to cause cancer cell death. A research program has been established with our collaborators at the University of Granada to investigate the changes in genetic and protein expression that occur in cancer cells as a consequence of being exposed to PRP. The objective of this work is to understand PRP on a molecular level changes in gene expression of the cancer cell post treatment. This will enable us to identify new, patentable drugs which we can develop such as synthetic recombinant proteins designed to improve the quality, safety and performance of pro-enzymes used in our current formulations.
Target Indications
The management of cancer differs widely, with a multitude of factors impacting on the choice of treatment strategy. Some of those factors include:
| · | the type of tumor, usually defined by the tissue in the body from which it originated; |
| · | the extent to which it has spread beyond its original location; |
| · | the availability of treatments, driven by multiple factors including cost, drugs approved, local availability of suitable facilities etc.; |
| · | regional and geographic differences; |
| · | whether the primary tumor is amenable to surgery, either as a potentially curative procedure, or as a palliative one; and |
| 17 |
| · | the balance between potential risks and potential benefits from the various treatments, and probably most importantly, the patient’s wishes. |
For many patients with solid cancers, such as breast, colorectal, lung and pancreatic cancer, surgery is frequently the first treatment option, frequently followed by first line chemotherapy +/- radiotherapy. Whilst hopefully such procedures are curative, in many instances the tumor returns, and second line treatment strategies are chosen in an effort to achieve a degree of control of the tumor. In most instances, the benefit is temporary, and eventually the point is reached where the patient’s tumor either fails to adequately respond to treatment, or the treatment has unacceptable toxicity which severely limits its usefulness.
Should the proposed Phase I, II and III clinical trials confirm the efficacy of our product candidates, along with the excellent safety and tolerability profile suggested by pre-clinical studies conducted, to date, our product will have utility in a number of clinical situations including:
| · | in the early stage management of solid tumors, most likely as part of a multi-pronged treatment strategy in combination with existing therapeutic interventions; |
| · | as a product that can be administered long term for patients following standard treatment approaches, such as surgery, or chemotherapy, in order to prevent or delay recurrence. |
| · | as a preventative measure for patients at risk of developing cancer based on genetic screening. |
In the near term, we plan to target patients with solid tumors, most likely colorectal and pancreatic tumors, for whom other treatment options have been exhausted. This is a common approach by which most new drugs for cancer are initially tested. Once efficacy and safety has been demonstrated in this patient population, exploration of the potential utility of the drug in earlier stage disease can be undertaken, together with investigation of the drug’s utility in other types of cancer.
Development Strategy
Our goal is to undertake early stage non-clinical and clinical development of our drug products through to a significant value inflexion point, where the commercial attractiveness of a drug in development, together with a greater likelihood of achieving market authorization, may attract potential interest from licensees seeking to acquire new products. Such value inflexion points in the context of cancer drugs are typically at the point where formal, controlled clinical trials have demonstrated either ‘efficacy’ or ‘proof of concept’ – typically meaning that there is controlled clinical trial evidence that the drug is effective in the proposed target patient population, has an acceptable safety profile, and is suitable for further development. From a ‘big picture’ perspective, it is our intention to progress the development of its technology through to completion of Phase I clinical trials and then to seek a licensee for further development beyond that point.
As part of that commercial strategy, we will:
| · | continue research and development to build our existing intellectual property portfolio, and to seek new, patentable discoveries; |
| · | seek to ensure all product development is undertaken in a manner that makes its products approvable in the major pharmaceutical markets, including the U.S., Europe, the UK and Japan; |
| · | aggressively pursue the protection of our technology through all means possible, including patents in all major jurisdictions, and potentially trade secrets; |
| · | make strategic acquisitions to acquire new companies that have products or services that complement our future goals. |
| 18 |
Development Plan and Milestones
PRP
We plan to progress PRP down a conventional non-clinical and early stage clinical development pathway either in Central, or Eastern Europe for:
| · | the manufacture of PRP for non-clinical development; |
| · | non-clinical safety toxicology studies; |
| · | regulatory approval to conduct a Phase I study in the relevant country, and submit it to the applicable regulatory authority for approval; and |
| · | a twelve month Phase I dose escalating study in cancer patients with advanced solid tumors who have failed one or more previous cancer therapies. |
We anticipate receiving the Phase IIa proof of concept milestone in approximately three years, subject to regulatory approval in Europe and the US, and the results from our research and development and licensing activities.
Our overheads are likely to increase from its current level as our lead product candidate, PRP progresses down the development pathway. This increase will be driven by the need to increase our internal resources in order to effectively manage our research and development activities.
We are initially seeking to raise sufficient capital to complete Phase I clinical trials over the next twenty-four months, although additional capital may be sought after twelve months to support expansion of research and development activities and our overheads (assuming planned expansion of internal resources are approved internally and completed accordingly).
Anticipated timelines
| 2014 | 2015 | 2016 | |||||||||||||||
| Q4 | Q1 | Q2 | Q3 | Q4 | Q1 | Q2 | Q3 | ||||||||||
| Complete animal efficacy models on PRP | X | ||||||||||||||||
| Manufacturing, production of drug substance and product for preclinical and clinical trials | X | X | X | ||||||||||||||
| Non-clinical development | X | X | X | ||||||||||||||
| Obtain regulatory approval | X | ||||||||||||||||
| Phase I | X | X | X | X | |||||||||||||
For the period from October to December 2014, we intend to complete animal efficacy models on PRP. We anticipate the cost of the studies to be $150,000.
For the period from January to September 2015, we intend to complete the manufacturing, production of drug substance and product for preclinical and clinical trials, as well as undertaking formal toxicology studies. We anticipate the cost to be $600,000 and $650,000, respectively.
For the period from September 2015 to September 2016, we intend to complete a Phase I study in advanced cancer patients with solid tumors and the anticipated costs will be $900,000 approximately.
| 19 |
Non Clinical Development
Cell line studies have been performed optimizing the ratio of the three components in our product, PRP. These studies demonstrate synergistic activity over the individual components. Additional preclinical efficacy and toxicology studies will be completed in the near future.
Bio-analytical assays for PRP will be developed prior to commencing the dose-range finding studies. In addition, the potential for E-cadherin to be used as a biomarker for PRP activity will be explored.
We are planning to develop PRP as an intravenous injection. Consequently, dose selection for GLP safety toxicology studies will be determined. Twenty – eight day GLP safety studies may also be necessary for PRP.
Clinical Development
It is proposed to perform the first-in-human study in advanced cancer patients with solid tumours. Typically these studies are dose-escalation studies aiming to identify the maximum tolerated dose (MTD). The study design will consist of single- or multiple-center, open-labelled, dose-escalating study in cancer patients with advanced solid tumours who have failed one or more previous therapies. Approximately 20 to 30 patients will be assessed, with the option to expand the MTD cohort to 10 patients. Dose limiting toxicities will be determined at the end of a one month cycle. Pharmacokinetic and exploratory pharmacodynamics data will be collected. Patients will remain on treatment until disease progression or the study finishes, at which time any remaining patients will be transferred to a long-term safety protocol.
If there is any patient with stable
disease or better, as defined by RECIST (Response Evaluation Criteria in Solid Tumors) and the safety profile is suitable then
PRP will progress to a second study.
POP1
As outlined previously, a research program has been established with our collaborators at the University of Granada to investigate the changes in genetic and protein expression that occur in cancer cells as a consequence of being exposed to our pro-enzyme formulation. The objective of this work is to understand at the molecular level the targets of our pro-enzyme formulation, thereby providing the opportunity for new, patentable drugs which can be developed further. We plan to commence a targeted drug discovery program utilizing the identified molecular target to search for novel anticancer agents.
Financial Objectives
Multiple factors, many of which are outside of Propanc’s control, can impact on the ability of Propanc to achieve its target objectives within the planned time and budgetary constraints. Subject to these caveats, it is Propanc’s objective to achieve the following R&D milestones within the proposed budget:
| · | PRP completed Phase I clinical trial. |
| · | Development candidate identified from the POP1 program. |
Corporate Strategy
We operate as a ‘virtual’ company contracting services, skills and expertise as required to achieve our scientific and corporate objectives. As the business grows and gains more personnel, outsourcing will continue to be the preferred model, where fixed and variable costs are carefully managed on a project by project basis. This means our research and development activities will be carried out by third parties. So far, we have engaged our research partners from the Universities of Bath and Granada. Additional third parties with specific expertise in research, compound screening and manufacturing (including raw material suppliers) will be contracted as required. Initial discussions have been held with several third parties and will be contracted as we progress into the next stages of the development process.
| 20 |
Our initial focus will be to organize, coordinate and finance the various parts of the drug development pipeline. New personnel will be carefully introduced into the company over a period of time as the company’s research and development activities expand. They will have specific expertise in product development, manufacture & formulation, regulatory affairs, toxicology, clinical operations and business development (including intellectual property management, licensing and other corporate activities).
In the first instance, additional clinical management and development expertise is likely to be required for our lead product therefore we anticipate an increase in employees in order to effectively manage our contractors as the project progress down the development pathway.
This outsourcing strategy is common in the biotechnology sector, and is an efficient way to obtain access to the necessary skills required to progress a project, in particular as the required skills change as the project progresses from discovery, through manufacturing and non-clinical development, and into clinical trials. We anticipate continuing to utilize this model, thereby retaining the flexibility to contract in the appropriate resource as and when required.
We intend to seek and identify potential licensing partners for our product candidates as they progress through the various development stages, reaching certain milestones and value inflection points. If a suitable licensee is identified, a potential licensing deal could consist of payments for certain milestones, plus royalties from future sales if the product is able to receive approval the relevant regulatory authorities where future product sales are targeted. We intend to seek and identify potential licensees based on the initial efficacy data from Phase I clinical trials within the next 18 to 24 months.
As part of our overall expansion strategy, we are investigating potential intellectual property acquisition opportunities to expand our product portfolio. Whilst the company’s initial focus is on the development of PRP as the lead product candidate, potential product candidates may also be considered for future preclinical and clinical development. These potential opportunities have arisen from other research and development organizations, which either own existing intellectual property, or currently developing new intellectual property, which may be of interest to us. These potential opportunities are potentially new cancer treatments which are potentially less toxic than existing treatment approaches and are able to fill an existing gap in the treatment process, such as a systemic de-bulking method which could reduce the size and threat of metastases to a more manageable level for late stage cancer patients. We believe these potential treatment approaches will be complementary to existing treatment regimens and our existing product candidate, PRP. No formal approaches have been made at this stage and it is unknown whether we will engage in this discussion in the near future. However, we remain hopeful that as PRP progresses further down the development pathway, future opportunities may arise to utilize the expertise of our management and scientific personnel for future prospective research and development projects.
Current Operations
We are at a pre-revenue stage. We do not know when, if ever, we will be able to commercialize our products. Presently, we are focusing our efforts on organizing, coordinating and financing the various aspects of the drug research and development program outlined earlier in this document. In order to commercialize our products, we must complete preclinical development, and Phase I, II and III clinical trials in Europe, the U.S., Australia, or elsewhere, and satisfy the applicable regulatory authority that PRP is safe and effective. We estimate that this will take approximately seven years. As described previously, when we have advanced our development projects sufficiently down the development pathway to achieve a major increase in value, such as obtaining interim efficacy data from Phase I clinical trials, we will seek a suitable licensing partner to complete the remaining development activities, obtain regulatory approval, and market the product.
| 21 |
Current Therapies/Drugs Available
We are developing a therapeutic solution for the treatment of patients with advanced stages of cancer targeting solid tumors, which is cancer that originates in organs or tissues other than bone marrow or the lymph system. Common cancer types classified as solid tumors include lung, colorectal, ovarian cancer, pancreatic cancer and liver cancers. In each of these indications, there is a large market opportunity to capitalize on the limitations of current therapies.
Current therapeutic options for the treatment of cancer offer, at most, a few months of extra life or tumor stabilization. Some experts believe that drugs that kill most tumor cells do not affect cancer stem cells which can regenerate the tumor (e.g. chemotherapy). Studies are revealing the genetic changes in cells that cause cancer and spur its growth this research is providing scientific researchers with dozens of potential “targets” for drugs. Tumor cells, however, can develop resistance to drugs.
Limitations of Current Therapies
PRP was developed because of the limitation of current cancer therapies. While surgery is often safe and effective for early stage cancer, many standard therapies for late stage cancer urgently need improvement; with current treatments generally providing modest benefits, and frequently causing significant adverse effects. Our focus is to provide oncologists and their patients with therapies for metastatic cancer which are more effective than current therapies, and which have a substantially reduced side effect profile.
While progress has been made within the oncology sector in developing new treatments, the overall cancer death rate has only improved 7% over the last 30 years. Most of these new treatments have some limitations, such as:
| · | significant toxic effects; |
| · | expense; and |
| · | limited survival benefits. |
We believe that our treatment will provide a competitive advantage over the following treatments:
| · | Chemotherapeutics: Side effects from chemotherapy can include pain, diarrhea, constipation, mouth sores, hair loss, nausea and vomiting, as well as blood-related side effects, which may include a low number of infection fighting white blood cell count (neutropenia), low red blood cell count (anemia), and low platelet count (thrombocytopenia). Our goal is to demonstrate that our treatment will be more effective than chemotherapeutic and hormonal therapies with fewer side effects. |
| · | Targeted therapies: Most common type is multi-targeted kinase inhibitors (molecules which inhibit a specific class of enzymes called kinases). Common side effects include fatigue, rash, hand–foot reaction, diarrhea, hypertension and dyspnoea (shortness of breath). Furthermore, tyrosine kinases inhibited by these drugs appear to develop resistance to inhibitors. Whilst the clinical findings with PRP are early and subject to confirmation in future clinical trials, no evidence has yet been observed of the development of resistance by the cancer to PRP. |
| · | Monoclonal antibodies: Development of monoclonal antibodies is often difficult due to safety concerns. Side effects which are most common include skin and gastro-intestinal toxicities. For example, several serious side effects from Avastin, an anti-angiogenic cancer drug, include gastrointestinal perforation and dehiscence (e.g. rupture of the bowel), severe hypertension (often requiring emergency treatment) and nephrotic syndrome (protein leakage into the urine). Antibody therapy can be applied to various cancer types in some cases, but can also be limited to certain genetic sub populations in many instances. |
| · | Immunotherapy: There is a long history of attempts to develop therapeutic cancer vaccines to stimulate the body’s own immune system to attack cancer cells. These products, whilst they generally do not have the poor safety profile of standard therapeutic approaches, have rarely been particularly effective. Whilst there are a number of therapeutic cancer vaccines currently in development, most are in the early stages of clinical development. To date, only one therapeutic cancer vaccine has been approved by the US Food and Drug Administration. |
| 22 |
Recent Development
Tarpon Bay Settlement Agreement
In July 2014, we entered into a Settlement and Stipulation Agreement (the “Settlement Agreement”) with Tarpon Bay Partners, LLC (“Tarpon”) to have Tarpon acquire certain portions of our liabilities to creditors (“Creditors”) in exchange for our obligation to issue shares of common stock to Tarpon, which shares of common stock would then be sold by Tarpon and 65% of the net proceeds, as defined, distributed to the Creditors. The shares are to be freely traded shares issued pursuant to section 3(a)(10) of the Securities Act of 1933.
Under the terms of the Settlement Agreement, the variable quantity of common stock would be issued in tranches such that the Tarpon would not own more than 9.99% of the outstanding shares of common stock at any time.
In connection to the Settlement Agreement, in May 2014, we also paid an expense fee of $25,000 in the form of a convertible promissory note.
Tarpon entered into agreements through July 2014 with the Creditors to acquire $627,998 in liabilities of the Company and filed a complaint with the Second Judicial Circuit Court in Leon County, Florida seeking a judgment against the Company for such amount. A court order based on this complaint was issued on September 9, 2014, (the "Court Order Date") resulting in the transfer of $627,998 in liabilities of the Company to the Tarpon. In addition, upon entry of the order, we became obligated to issue to Tarpon a purchaser fee of $50,000 worth of common stock priced at 75% of the average closing bid prices for the 10 days immediately preceding the date of the order. As a result of the purchased liabilities and purchaser fee, we became obligated to issue to the purchaser approximately $1,034,000 worth of common stock. These liabilities now meet the criteria of stock settled debt under ASC 480 resulting in the recording of a liability premium of approximately $356,000 with a charge to interest expense on the court order date.
We issued an initial tranche of 7,426,000 shares of common stock to Tarpon in September 2014.
Issuance of Convertible Promissory Notes
On May 29 and May 30, 2014, we entered into securities purchase agreements with Union Capital, LLC, LG Capital Funding, LLC and Adar Bays, LLC pursuant to which we executed a total of six (6) convertible promissory notes with these purchasers resulting in aggregate loan amount of $200,000. We agreed to pay 8% interest per annum on the principal amount and the maturity dates are May 29 and May 30, 2015. The notes are convertible at the option of the holder at any time after 180 days at a rate of 55% of the lowest trading bid price of the our common stock for the ten (10) prior trading days including the date upon which the conversion notice was received. Proceeds from the financing have been used primarily to support working capital expenses such as accounting, legal, intellectual property and administrative expenses.
| 23 |
Southridge Partners Financing
On July 18, 2014, we entered into an equity purchase agreement (the “EPA”) with Southridge Partners II, L.P. (“Southridge”) pursuant to which, the investor shall, from time to time over the 24-month term of the agreement, commit to purchase up to $5,000,000 of the company’s common stock in connection to put notices by the company. The purchase price of the common stock under the EPA is equal to ninety percent to the lowest closing bid price, quoted by the exchange or principal market company’s common stock is traded on, on any trading day during the ten (10) trading days immediately after the company delivers to Southridge a put notice in writing requiring Southridge to purchase shares of the Company subject to certain terms of the EPA.
In connection with the execution of the EPA, on the same date, we also entered into a registration rights agreement (the “Registration Rights Agreement”) with Southridge. Pursuant to the Registration Rights Agreement, we agreed to have an initial registration statement declared effective within a certain time frame. The mechanics triggering the issuance of those securities were fully negotiated and set forth in the EPA disclosed previously with the SEC.
Also in connection with the execution of the EPA, on the same date, we executed a promissory note to Southridge resulting in a loan amount of $50,000 bearing an interest rate of 0% per annum, with the maturity date ofJanuary 31, 2015.
Market Opportunity
Total global oncology drug sales reached $91 billion in 2013 and are growing at 5% annually. In particular, targeted therapies have significantly increased their share from 11% a decade ago to 46% last year. Biological products, which are products often made from natural resources, such as human, animal and microorganisms, represent nearly half of the oncology market. More recently, new drug launches have concentrated on small molecules, including kinase inhibitors. However, these new drugs cost more because they are meant for smaller patient populations.
Our cancer treatment is intended to be positioned among the five types of cancer drug classes currently contributing to the significant growth in the oncology market. The five main drug classes are chemotherapeutics, hormonals, immunotherapy and vaccines, targeted therapies and monoclonal antibodies.
Demand for new cancer products can largely be attributed to a combination of a rapidly aging population in western countries and changing environmental factors, which together are resulting in rising cancer incidence rates. According to the World Health Organization, all cancers (excluding non-melanoma skin cancer) are expected to increase from 8.2 million annual deaths in 2012 to over 10 million annual deaths by 2020, exceeding 13 million annual deaths by 2030. As such, global demand for new cancer treatments which are effective, safe and easy to administer is rapidly increasing. Our treatment will potentially target many aggressive tumor types for which little or few treatment options exist.
We plan to target patients with solid tumors, most likely colorectal and pancreatic tumors, for whom other treatment options have been exhausted. Globally these cancers resulted in over 694,000 deaths per year in 2012. With such a high mortality rate, a substantial unmet medical need exists for new treatments.
For example, current standard treatment for colorectal cancer consists of cytotoxics, which are associated with high levels of toxicity. Despite the relatively recent approval of Erbitux™ and Avastin™, both of which are monoclonal antibodies, for the treatment of colorectal cancer, significant treatment-related adverse effects continue to be problematic for patients with colorectal cancer. The need exists for tolerable agents that will improve quality of life for patients as well as offering a potential cure (Datamonitor, 2004).
For pancreatic cancer, there is a lack of effective therapies on the market for pancreatic cancer and any newly approved agents with some efficacy are likely to see significant uptake once commercialized (Datamonitor, 2004). Targeted therapies may fulfill this need, although further intensive research and development is necessary.
| 24 |
Once the efficacy and safety of PRP has been demonstrated in late stage patient populations, we plan to undertake exploration of the utility of the drug in earlier stage disease, together with investigation of the drug’s utility in other types of cancer.
Anticipated Market Potential
It is difficult to estimate the size of the market opportunity for this specific type of product as a clinically proven, pro-enzyme formulated suppository marketed to oncologists across global territories for specific cancer indications, to the best of management’s knowledge, has not been previously available.
However, the markets for potential market for colorectal and pancreatic cancer may be characterized as follows:
| · | Colorectal cancer: In 2011, according to available information online, the global colorectal cancer therapeutics market was worth $8.3 billion. The market is expected to decrease marginally to $7.8 billion by 2021 because of generic competition for a key cytotoxic agent, oxaliplatin, as well as the entry of biosimilar competitors for key targeted biological agents. Therefore, demand for new and innovative treatment approaches will be significant to support future growth and continue to improve treatment standards. |
| · | Pancreatic cancer: The world market for pancreatic cancer drugs is projected to grow to $1.63 billion by the year 2017. This rapid market expansion is due to launch of Celgene’s Abraxane in the US and Europe in 2013 and 2014. Abraxane will represent nearly 60% of the cancer therapeutics market by the end of 2017. |
Based on the current situation for these two markets, we believe there is an attractive opportunity in both the colorectal and pancreatic cancer market sectors for the introduction of a clinically proven product which can achieve new benefits for patients in terms of survival and quality of life. The current concentration of products suggests oncologists may be willing to try newly approved products, particularly if they can exhibit a favorable safety profile, although substantive R&D activities will be necessary to both obtain regulatory approval, and to generate the clinical safety and efficacy data needed to convince clinicians to use a new product.
License Agreements
We previously sponsored a collaborative research project at Bath University to investigate the cellular and molecular mechanisms underlying the potential clinical application of our proprietary pro-enzyme formulation.
As a result of this undertaking, we entered into a Commercialization Agreement with Bath University, dated 12 November 2009, where, initially, Propanc, held an exclusive license with the University of Bath (UK), where we, and the University, co-own the intellectual property relating our pro-enzyme formulations. The Commercialization Agreement provides for Bath to assign the Patents to Propanc in certain specified circumstances, such as successful completion of a Phase I clinical trial and commencement of a Phase IIa (Proof of Concept) clinical trial.
On June 14 2012, Propanc and Bath University agreed to an earlier assignment of the patents pursuant to an Assignment and Amendment Deed, on the proviso that Bath retains certain rights arising from the Commercialization Agreement, as follows:
| · | Bath reserves for itself (and its employees and students and permitted academic sub-licensees regarding Research Use) the non-exclusive, irrevocable, worldwide, royalty free right to use the Patents for Research Use. |
| 25 |
| · | The publication rights of Bath specified in the contract relating to the Original Research made between the Parties with an effective date of 18th July 2008 shall continue in force. |
| · | Propanc shall pay to the University of Bath a royalty being two (2) per cent of any and all net revenues. |
| · | Propanc shall use all reasonable endeavors to develop and commercially exploit the Patents for the mutual benefit of Bath and Propanc to the maximum extent throughout the Territory in the Field and in each Additional Field and to obtain, maintain and/or renew any licenses or authorizations which are necessary to enable such development and commercial exploitation. Without prejudice to the generality of the foregoing, Propanc shall comply with all relevant regulatory requirements in respect of its sponsoring and/or performing clinical trials in man involving the administration of a product or materials within a claim of the Patents. |
| · | Propanc shall take out with a reputable insurance company and maintain liability insurance cover prior to the first human trials. |
We intend to work together with the University of Bath to patent and commercialize these discoveries, while continuing to elucidate the properties of pro-enzymes with the long-term aim of screening new compounds for development. At present, we are engaged in discussions with several technology companies who are progressing new developments in the oncology field as potential additions to our product line. Initially targeting the oncology sector, our focus is to identify and develop novel treatments which are highly effective targeted therapies, with few side effects as a result of toxicity to healthy cells.
Intellectual Property
We have filed an international patent application directed to enhance pro-enzyme formulations and combination therapies comprising trypsinogen and chymotrypsin, and/or a number of other specific anti-cancer agents. The international patent application has been based on previous provisional patent applications filed by us capturing ongoing research and development in this area.
The international patent application was filed on October 22, 2010, which claims priority for Australian provisional patent application nos. 2009905147 (filed October 22, 2010) and 2010902655 (filed June 17, 2010).
The details of such patent are as follows:
| · | Title: A Pharmaceutical Composition For Treating Cancer Comprising Trypsinogen And/Or Chymotrypsinogen And An Active Agent Selected From A Selenium Compound, A Vanilloid Compound, And A Cytoplasmic Glycolysis Reduction Agent. |
| · | Date filed: 22nd October 2010. |
| · | Jurisdiction: The Patent Cooperation Treaty or PCT is an international agreement for filing patent applications having effect in up to 117 countries. Under the PCT, an inventor can file a single international patent application in one language with one patent office in order to simultaneously seek protection for an invention in up to 117 countries. |
We recently completed the 30-month national phase filing deadline for this international PCT application and has commenced entering the national phase in individual countries and regions, including United States, Canada, Japan, Brazil, China, Mexico, Hong Kong, Singapore, Israel, Chile, Peru, Malaysia, Vietnam, Indonesia, Europe, Russia, India, Australia and South Korea. The patent is granted in South Africa and New Zealand. Further, provisional patents are also expected to be filed to capture and protect additional patentable subject matter that is identified, namely further enhanced formulations, combination treatments, use of recombinant products, modes of action and molecular targets.
Our intellectual property portfolio also includes an extensive amount of confidential information, know-how and expertise in relation to the development and formulation of our pro-enzyme based combination therapies.
| 26 |
The basis of our intellectual property protection will be built around the following elements:
| · | Method of use: Understanding the mechanism of action of the PRP pro-enzyme formulations, enabling the identification of new molecular targets, potential new therapeutic compounds and identification of new formulations that are adapted to enhance activity. |
| · | Formulation: We have developed an enhanced formulation containing the pro-enzyme trypsinogen in combination with at least one of two types of identified compounds considered effective for providing synergistic enhancement of the pro-enzyme based formulations. A patentability assessment, based on an international prior art search, has indicated that strong potential exists for successfully obtaining patent claims covering the formulation. |
| · | Composition of Matter: Synthetic recombinant proteins designed to improve the quality, safety and performance of pro-enzymes used in the proposed formulations form part of the research and development program. |
Regulatory Issues
United States
Government oversight of the pharmaceutical industry is usually classified into pre-approval and post-approval categories. Most of the therapeutically significant innovative products marketed today are the subject of New Drug Applications (“NDA”). Preapproval activities, based on these detailed applications, are used to assure the product is safe and effective before marketing. In the United States, The Center for Drug Evaluation and Research, or CDER, is the Food and Drug Administration (the “FDA”) organization responsible for over-the-counter and prescription drugs, including most biological therapeutics, and generic drugs.
Before approval, the FDA may inspect and audit the development facilities, planned production facilities, clinical trials, institutional review boards, and laboratory facilities in which the product was tested in animals. After the product is approved and marketed, the FDA uses different mechanisms for assuring that firms adhere to the terms and conditions of approval described in the application and that the product is manufactured in a consistent and controlled manner. This is done by periodic unannounced inspections of production and quality control facilities by FDA’s field investigators and analysts.
Federal Food, Drug and Cosmetic Act and Public Health Service Act
Prescription drug and biologic products are subject to extensive pre- and post-market regulation by the FDA, including regulations that govern the testing, manufacturing, safety, efficacy, labelling, storage, record keeping, advertising and promotion of such products under the Federal Food, Drug and Cosmetic Act, the Public Health Service Act, and their implementing regulations. The process of obtaining FDA approval and achieving and maintaining compliance with applicable laws and regulations requires the expenditure of substantial time and financial resources. Failure to comply with applicable FDA or other requirements may result in refusal to approve pending applications, a clinical hold, warning letters, civil or criminal penalties, recall or seizure of products, partial or total suspension of production or withdrawal of the product from the market. FDA approval is required before any new drug or biologic, including a new use of a previously approved drug, can be marketed in the United States. All applications for FDA approval must contain, among other things, information relating to safety and efficacy, stability, manufacturing, processing, packaging, labelling and quality control.
New Drug Applications (NDAs)
The FDA’s NDA approval process generally involves:
| · | Completion of preclinical laboratory and animal testing in compliance with the FDA’s good laboratory practice, or GLP, regulations; |
| 27 |
| · | Submission to the FDA of an investigational new drug (“IND”) application for human clinical testing, which must become effective before human clinical trials may begin in the United States; |
| · | Performance of adequate and well-controlled human clinical trials to establish the safety, purity and potency of the proposed product for each intended use; |
| · | Satisfactory completion of an FDA pre-approval inspection of the facility or facilities at which the product is manufactured to assess compliance with the FDA’s “current good manufacturing practice” (“CGMP”) regulations; and |
| · | Submission to and approval by the FDA of a NDA. |
The preclinical and clinical testing and approval process requires substantial time, effort and financial resources, and Propanc cannot guarantee that any approvals for our product candidates will be granted on a timely basis, if at all. Preclinical tests include laboratory evaluation of toxicity and immunogenicity in animals. The results of preclinical tests, together with manufacturing information and analytical data, are submitted as part of an IND application to the FDA. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions about the conduct of the clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin. Our submission of an IND may not result in FDA authorization to commence clinical trials. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development. Further, an independent institutional review board (“IRB”), covering each medical centre proposing to conduct clinical trials must review and approve the plan for any clinical trial before it commences at that center and it must monitor the study until completed. The FDA, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive “good clinical practice” (“GCP”) regulations, which include requirements that all research subjects provide informed consent and that all clinical studies be conducted under the supervision of one or more qualified investigators.
For purposes of an NDA submission and approval, human clinical trials are typically conducted in the following sequential phases, which may overlap:
| · | Phase I: Trials are initially conducted in a limited population to test the product candidate for safety and dose tolerance. |
| · | Phase II: Trials are generally conducted in a limited patient population to identify possible adverse effects and safety risks, to determine the initial efficacy of the product for specific targeted indications and to determine dose tolerance and optimal dosage. Multiple Phase II clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more extensive Phase III clinical trials. |
| · | Phase III: These are commonly referred to as pivotal studies. When Phase II evaluations demonstrate that a dose range of the product is effective and has an acceptable safety profile, Phase III clinical trials are undertaken in large patient populations to further evaluate dosage, to provide substantial evidence of clinical efficacy and to further test for safety in an expanded and diverse patient population at multiple, geographically-dispersed clinical trial sites. Generally, replicate evidence of safety and effectiveness needs to be demonstrated in two adequate and well-controlled Phase III clinical trials of a product candidate for a specific indication. These studies are intended to establish the overall risk/benefit ratio of the product and provide adequate basis for product labelling. |
| · | Phase IV: In some cases, the FDA may condition approval of a NDA on the sponsor’s agreement to conduct additional clinical trials to further assess the product’s safety, purity and potency after NDA approval. Such post-approval trials are typically referred to as Phase IV clinical trials. |
Progress reports detailing the results of the clinical studies must be submitted at least annually to the FDA and safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events. Concurrent with clinical studies, sponsors usually complete additional animal studies and must also develop additional information about the product and finalize a process for manufacturing the product in commercial quantities in accordance with CGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things; the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Moreover, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
| 28 |
The results of product development, preclinical studies and clinical trials, along with the aforementioned manufacturing information, are submitted to the FDA as part of a NDA. NDA’s must also contain extensive manufacturing information. Under the Prescription Drug User Fee Act, or PDUFA, the FDA agrees to specific goals for NDA review time through a two-tiered classification system, Standard Review and Priority Review. Standard Review is applied to products that offer at most, only minor improvement over existing marketed therapies. Standard Review NDAs have a goal of being completed within a ten-month timeframe, although a review can take a significantly longer amount of time. A Priority Review designation is given to products that offer major advances in treatment, or provide a treatment where no adequate therapy exists. A Priority Review means that the time it takes the FDA to review a NDA is six months. It is likely that our product candidates will be granted Standard Reviews. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA may refer the application to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations.
The FDA may deny approval of a NDA if the applicable regulatory criteria are not satisfied, or it may require additional clinical data or additional pivotal Phase III clinical trials. Even if such data are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than Propanc do. Once issued, product approval may be withdrawn by the FDA if ongoing regulatory requirements are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing, including Phase IV clinical trials, Risk Evaluation and Mitigation Strategies, or REMS, and surveillance programs to monitor the effect of approved products that have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs. Products may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are any modifications to the drug, including changes in indications, labelling or manufacturing processes or facilities, approval of a new or supplemental NDA may be required, which may involve conducting additional preclinical studies and clinical trials.
Other U.S. Regulatory Requirements
After approval, products are subject to extensive continuing regulation by the FDA, which include company obligations to manufacture products in accordance with GMP, maintain and provide to the FDA updated safety and efficacy information, report adverse experiences with the product, keep certain records and submit periodic reports, obtain FDA approval of certain manufacturing or labelling changes, and comply with FDA promotion and advertising requirements and restrictions. Failure to meet these obligations can result in various adverse consequences, both voluntary and FDA-imposed, including product recalls, withdrawal of approval, restrictions on marketing, and the imposition of civil fines and criminal penalties against the NDA holder. In addition, later discovery of previously unknown safety or efficacy issues may result in restrictions on the product, manufacturer or NDA holder.
Propanc, and any manufacturers of our products, are required to comply with applicable FDA manufacturing requirements contained in the FDA’s GMP regulations. GMP regulations require among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation. The manufacturing facilities for our products must meet GMP requirements to the satisfaction of the FDA pursuant to a pre-approval inspection before Propanc can use them to manufacture our products. Propanc, and any third-party manufacturers, are also subject to periodic inspections of facilities by the FDA and other authorities, including procedures and operations used in the testing and manufacture of our products to assess our compliance with applicable regulations.
| 29 |
With respect to post-market product advertising and promotion, the FDA imposes a number of complex regulations on entities that advertise and promote pharmaceuticals, which include, among others, standards for direct-to-consumer advertising, promoting products for uses or in patient populations that are not described in the product’s approved labelling (known as “off-label use”), industry-sponsored scientific and educational activities, and promotional activities involving the Internet. Failure to comply with FDA requirements can have negative consequences, including adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties. Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such off-label uses.
Changes to some of the conditions established in an approved application, including changes in indications, labelling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented. A NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing NDAs.
Adverse event reporting and submission of periodic reports is required following FDA approval of a NDA. The FDA also may require post-marketing testing, known as Phase IV testing, risk mitigation strategies and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product.
European Union
In addition to regulations in the United States, Propanc will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products. Whether or not Propanc obtains FDA approval for a product, Propanc must obtain approval of a product by the comparable regulatory authorities of foreign countries before Propanc can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials product licensing, pricing and reimbursement vary greatly from country to country.
Under European Union regulatory systems, Propanc must submit and obtain authorization for a clinical trial application in each member state in which Propanc intend to conduct a clinical trial. After Propanc have completed clinical trials, Propanc must obtain marketing authorization before Propanc can market its product. Propanc must submit applications for marketing authorizations for oncology products under a centralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. The European Medicines Agency (the “EMA”) is the agency responsible for the scientific evaluation of medicines that are to be assessed via the centralized procedure.
Australia
In Australia, the relevant regulatory body responsible for the pharmaceutical industry is the Therapeutics Goods Administration (the “TGA”). Prescription medicines are regulated under the Therapeutic Goods Act 1989. Under the Therapeutic Goods Act, the Therapeutic Goods Administration evaluates new products for quality, safety and efficacy before being approved for market authorization, according to similar standards employed by the FDA and EMA in the United States and European Union, respectively. However, receiving market authorization in one or two regions does not guarantee approval in another.
Third-Party Payor Coverage and Reimbursement
Although none of our product candidates have been commercialized for any indication, if they are approved for marketing, commercial success of our product candidates will depend, in part, upon the availability of coverage and reimbursement from third-party payors at the federal, state and private levels.
| 30 |
Employees
Under European Union regulatory systems, Propanc must submit and obtain authorization for a clinical trial application in each member state in which Propanc intend to conduct a clinical trial. After Propanc have completed clinical trials, Propanc must obtain marketing authorization before Propanc can market its product. Propanc must submit applications for marketing authorizations for oncology products under a centralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. The European Medicines Agency (the “EMA”) is the agency responsible for the scientific evaluation of medicines that are to be assessed via the centralized procedure.
Our Corporate Information
Our principal executive office is located at Level 13, Suite 1307, 530 Little Collins Street, Melbourne, VIC, 3000, Australia and our phone number is +61 (0) 3 9614 2795. Our Australian subsidiary, Propanc PTY Ltd. shares the same office with us.
Research and Development
During the last two completed fiscal years ending June 30, 2014 and 2013, we have spent $8,168 and $12,344, respectively, on research and development expenses.
Available Information
Copies of our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and other documents that we will file with or furnish to the SEC will be available free of charge by sending a written request to our Corporate Secretary at our corporate headquarters. Additionally, the documents we file with the SEC is or will be available free of charge at the SEC’s Public Reference Room at 100 F Street, NE, Washington D.C. 20549. Other information on the operation of the Public Reference Room is or will be available by calling the SEC at (800) SEC-0330.
Smaller reporting companies are not required to provide the information required by this item.
Item 1B. Unresolved Staff Comments.
None.
Our corporate office is located at Level 13, Suite 1307, 530 Little Collins Street, Melbourne, VIC, 3000, Australia. The lease costs $942 per month and expires on one month’s notice by either Propanc or the leasing company.
From time to time, we may be involved in litigation in the ordinary course of business.
We are currently not involved in any litigation that we believe could have a material adverse effect on our financial condition or results of operations. To our knowledge, there is no action, suit, proceeding, inquiry or investigation before or by any court, public board, government agency, self-regulatory organization or body pending or, to the knowledge of our executive officers or any of our subsidiaries, threatened against or affecting our company, our common stock, any of our subsidiaries or any of our companies or our companies’ subsidiaries’ officers or directors in their capacities as such, in which an adverse decision could have a material adverse effect.
Item 4. Mine Safety Disclosures.
Not applicable.
| 31 |
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Our common stock has been quoted on the OTC Bulletin Board since June 15, 2012 and is currently quoted under the symbol “PPCH”.
The following table sets forth the high and low bid quotations for our common stock as reported on the OTC Bulletin Board for the periods indicated.
| High
Bid* ($) | Low
Bid* ($) | |||||||
| 2014 | ||||||||
| Fourth quarter | $ | 0.47 | 0.08 | |||||
| Third quarter | $ | 0.10 | 0.10 | |||||
| Second quarter | $ | 0.20 | 0.10 | |||||
| First quarter | $ | 0.20 | 0.20 | |||||
| 2013 | ||||||||
| Fourth quarter | $ | 1.00 | 0.20 | |||||
| Third quarter | $ | 1.01 | 0.20 | |||||
| Second quarter | $ | 1.52 | 0.75 | |||||
| First quarter | $ | N/A | N/A | |||||
* The quotations of the closing prices reflect inter-dealer prices, without retail mark-up, markdown or commission.
Holders
As of October 14, 2014, there were 61 holders of record of our Common Stock.
Dividend Policy
We have not paid cash dividends on our common stock and do not plan to pay such dividends in the foreseeable future. Our board of directors (the “Board”) will determine our future dividend policy on the basis of many factors, including results of operations, capital requirements, and general business conditions. Dividends, under Delaware General Corporation Law, may only be paid from our net profits or surplus. To date, we have not had a fiscal year with net profits and do not have surplus.
Recent Sales of Unregistered Securities
In addition to those sales of unregistered securities previously disclosed in reports filed with the SEC during the fiscal year ended June 30, 2014, we issued the following securities without registration under the Securities Act of 1933.
On September 30, 2013 the Company’s subsidiary issued a debenture for $139,683 (AUD$150,000) plus warrants for 3,000,000 common shares of the Company. The Company agreed to pay 12% interest on the principal amount and the maturity date is December 31, 2015. The debenture is convertible only at the Company’s option into common stock at $0.0698 per share and is convertible at that same rate by the lender only upon default by the Company, as defined in the debenture.
| 32 |
On May 29 and May 30, 2014 the Company issued six convertible promissory notes to certain investors for an aggregate amount of $400,000, $200,000 of which are paid for by an offsetting $200,000 promissory notes issued to the Company by the investors. The Company agreed to pay 8% interest on the principal amount and the maturity date is one year from the execution date of the notes. The notes are convertible into company’s common stock at any time after the requisite Rule 144 holding period, subject to certain terms and conditions, at a conversion price equal to 55% of the lowest trading bid price in the ten (10) trading days prior to the conversion.
Outstanding Equity Awards
There are no outstanding equity awards.
Equity Compensation Plan Information
We currently do not have an equity compensation plan.
Director Compensation
We do not pay cash compensation to our directors for service on our Board and our employees do not receive compensation for serving as members of our Board. Directors are reimbursed for reasonable expenses incurred in attending meetings and carrying out duties as board members.
Item 6. Selected Financial Data.
Not applicable to smaller reporting companies.
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
FORWARD-LOOKING STATEMENTS
The information set forth in this Management's Discussion and Analysis of Financial Condition and Results of Operations (“MD&A”) contains certain “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, Section 21E of the Securities Exchange Act of 1934, as amended, and the Private Securities Litigation Reform Act of 1995, including, among others (i) expected changes in Propanc Health Group Corporation's (referred to herein as the "Company", or "Propanc", "we", "our", "ours" and "us") revenues and profitability, (ii) prospective business opportunities and (iii) our strategy for financing its business. Forward-looking statements are statements other than historical information or statements of current condition. Some forward-looking statements may be identified by use of terms such as “believes”, “anticipates”, “intends” or “expects”. These forward-looking statements relate to our plans, objectives and expectations for future operations. Although we believe that our expectations with respect to the forward-looking statements are based upon reasonable assumptions within the bounds of our knowledge of our business and operations, in light of the risks and uncertainties inherent in all future projections, the inclusion of forward-looking statements in this Annual Report should not be regarded as a representation by us or any other person that our objectives or plans will be achieved.
We assume no obligation to update these forward-looking statements to reflect actual results or changes in factors or assumptions affecting forward-looking statements.
You should read the following discussion and analysis in conjunction with the Financial Statements and Notes attached hereto, and the other financial data appearing elsewhere in this Annual Report.
| 33 |
Our revenues and results of operations could differ materially from those projected in the forward-looking statements as a result of numerous factors, including, but not limited to, the following: the risk of significant natural disaster, the inability of the Company to insure against certain risks, inflationary and deflationary conditions and cycles, currency exchange rates, and changing government regulations domestically and internationally affecting our products and businesses.
US Dollars are denoted herein by “USD”, "$" and "dollars".
Overview
Propanc Health Group Corporation, formerly Propanc PTY Ltd., ("the Company", "we", "us", "our") was incorporated in Melbourne, Victoria Australia on October 15, 2007, and is based in Melbourne, Victoria Australia.
On November 23, 2010, Propanc Health Group Corporation was incorporated in the state of Delaware. In January 2011, Propanc Health Group Corporation acquired all of the outstanding shares of Propanc PTY Ltd. on a one-for-one basis making it a wholly-owned subsidiary.
We are a research and development company whose primary activity is to develop new treatments for chronic diseases, in particular cancer. We have generated very limited revenue, have no cancer treatment products available to market and have no products which have reached the clinical trial stage. We require substantial additional financing to develop our products.
Critical Accounting Estimates
Below the Company will provide a discussion of its more subjective accounting estimation processes for purposes of (i) explaining the methodology used in calculating the estimates, (ii) the inherent uncertainties pertaining to such estimates, and (iii) the possible effects of a significant variance in actual experience, from that of the estimate, on the Company’s financial condition. Estimates involve the employ of numerous assumptions that, if incorrect, could create a material adverse impact on the Company’s results of operations and financial condition.
Foreign Currency Translation and Comprehensive Income (Loss): The Company’s functional currency is the Australian dollar (AUD). For financial reporting purposes, the Australian dollar has been translated into United States dollars ($) and/or USD as the reporting currency. Assets and liabilities are translated at the exchange rate in effect at the balance sheet date. Revenues and expenses are translated at the average rate of exchange prevailing during the reporting period. Equity transactions are translated at each historical transaction date spot rate. Translation adjustments arising from the use of different exchange rates from period to period are included as a component of stockholders’ equity (deficit) as “accumulated other comprehensive income (loss).” Gains and losses resulting from foreign currency transactions are included in the statement of operations and comprehensive loss as other income (expense).
Accounting for Income Taxes: The Company is governed by Australia and United States income tax laws, which are administered by the Australian Taxation Office and the United States Internal Revenue Service, respectively. The Company follows FASB ASC 740 when accounting for income taxes, which requires an asset and liability approach to financial accounting and reporting for income taxes. Deferred income tax assets and liabilities are computed annually for temporary differences between the financial statements and tax bases of assets and liabilities that will result in taxable or deductible amounts in the future based on enacted tax laws and rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized. Income tax expense is the tax payable or refundable for the period plus or minus the change during the period in deferred tax assets and liabilities.
| 34 |
The Company adopted provisions of ASC 740, Sections 25 through 60, “Accounting for Uncertainty in Income Taxes." These sections provide detailed guidance for the financial statement recognition, measurement and disclosure of uncertain tax positions recognized in the financial statements. Tax positions must meet a “more-likely-than-not” recognition threshold at the effective date to be recognized upon the adoption of ASC 740 and in subsequent periods.
Accounting for Stock Based Compensation: The Company records stock based compensation in accordance with ASC section 718, “Stock Compensation” and Staff Accounting Bulletin (SAB) No. 107 (SAB 107) issued by the Securities and Exchange Commission (SEC) in March 2005 regarding its interpretation of ASC 718. ASC 718 requires the fair value of all stock-based employee compensation awarded to employees to be recorded as an expense over the related requisite service period. The statement also requires the recognition of compensation expense for the fair value of any unvested stock option awards outstanding at the date of adoption. The Company values any employee or non-employee stock based compensation at fair value using the Black-Scholes Option Pricing Model.
The Company accounts for non-employee share-based awards in accordance with the measurement and recognition criteria of ASC 505-50 "Equity-Based Payments to Non-Employees.
Derivative Instruments
ASC Topic 815, Derivatives and Hedging (“ASC Topic 815”), establishes accounting and reporting standards for derivative instruments and for hedging activities by requiring that all derivatives be recognized in the balance sheet and measured at fair value. Gains or losses resulting from changes in the fair value of derivatives are recognized in earnings or recorded in other comprehensive income (loss) depending on the purpose of the derivatives and whether they qualify and have been designated for hedge accounting treatment. The Company does not have any derivative instruments for which it has applied hedge accounting treatment.
Research and Development Tax Credits: The Company may apply for Research and Development tax concessions with the Australian Taxation Office on an annual basis. Although the amount is possible to estimate at year end, the Australian Taxation Office may reject or materially alter the claim amount. Accordingly, the Company does not recognize the benefit of the claim amount until cash receipt since collectability is not certain until such time. The tax concession is a refundable credit. If the Company has net income then the Company can receive the credit which reduces its income tax liability. If the Company has net losses then the Company may still receive a cash payment for the credit, however, the Company's net operating loss carry forwards are reduced by the gross equivalent loss that would produce the credit amount when the income tax rate is applied to that gross amount. The concession is recognized as an income tax benefit, in operations, upon receipt.
Recent Accounting Pronouncements
Financial Accounting Standards Board, Accounting Standard Updates which are not effective until after June 30, 2014 are not expected to have a significant effect on the Company’s consolidated financial position or results of operations.
Results of Operations
The following discussion should be read in conjunction with the consolidated financial statements and notes thereto included elsewhere in this form 10-K. The results discussed below are of the Company and its wholly-owned Australian subsidiary, Propanc Pty Ltd.
For the Year Ended June 30, 2014 compared to the Year ended June 30, 2013
Revenue
For the fiscal years 2014 and 2013, we generated no revenue because the company is currently undertaking research and development activities for market approval and there were no sales generated in this period.
Administration Expense
Administration expense decreased to $742,037 for the year ended June 30, 2014 as compared with $1,336,482 for the year ended June 30, 2013. This decrease is primarily attributable to stock based expenses of $888,200 incurred during the year ending June 30, 2013.
Occupancy Expense
Occupancy expense decreased by $2,058 to $11,016 for the year ended June 30, 2014.
| 35 |
Research and Development Expenses
Research and Development was $8,168 for the year ended June 30, 2014 as compared with $12,344 for the year ended June 30, 2013. Research and Development expenditure has been minimal over the past 24 months, including the last 12 months, as the Company raises sufficient capital to undertake its next stage of development for its current programs. The Company and its Directors continue to expend its efforts to continue creating value by completing its patent filings and publishing its scientific discoveries, and is negotiating with third parties to assist with raising the capital needed to complete its planned research and development activities.
Interest Expense/Income
Interest expense increased to $93,147 for the year ended June 30, 2014 as compared with $5,465 for the year ended June 30, 2013. Interest expense is comprised of $16,931 face interest, $49,029 debt discount amortization, and $27,187 accretion of debt premium. This increase is primarily attributable to interest bearing loans made to the company during the fiscal year.
Income Tax Benefit
During the years-ended ended June 30, 2014 and 2013, the Company applied for and received from the Australian Taxation Office a research and development tax credit in the amount of $48,267 and $60,461.
Net loss
Net loss decreased to $829,564 for the year ended June 30, 2014 as compared with $1,442,638 for the year ended June 30, 2013. The decrease is primarily attributable to administrative expenses which decreased by $594,445, for the year ended June 30, 2014.
Liquidity and Capital Resources
| For the Fiscal Year Ended June 30, | ||||||||
| 2014 | 2013 | |||||||
| Net cash used in operating activities | $ | (226,442 | ) | $ | (128,647 | ) | ||
| Net cash used in investing activities | $ | - | $ | - | ||||
| Net cash provided by financing activities | $ | 311,141 | $ | 128,465 | ||||
Net cash used in operations was $128,647 for the fiscal year ended June 30, 2013 compared to $226,442 for the same period in 2014. This decrease was primarily attributable to less activity due to the company's limited cash resources.
There were no cash transactions from investing activities in fiscal year 2014 or 2013.
Cash flows provided by financing activities for the fiscal year ended June 30, 2014 were $311,141 compared to $128,465 for the fiscal year ended June 30, 2013. In 2014 we had loan proceeds from officers and directors of $72,158 and from unrelated parties of $10,542, and we issued $340,000 worth of convertible promissory notes with net proceeds after costs of $273,959, while in 2013 we had loan proceeds from officers and directors of $123,814 and from unrelated parties of $37,506.
We have substantial capital resource requirements and have incurred significant losses since inception. As of June 30, 2014, we had $87,799 in cash. Based upon our current business plans, we will need considerable cash investments to be successful. Such capital requirements are in excess of what we have in available cash and what we currently have commitment for. Therefore, we do not have enough available cash to meet our obligations over the next 12 months.
| 36 |
Related Party Transactions
Since inception, Propanc Health Group Corporation has conducted transactions with directors and director related entities. These transactions included the following:
As of June 30, 2014 and 2013, the Company owed certain directors a total of $161,975 and $130,689 respectively, for money loaned to the Company throughout the years.
As of June 30, 2014 and 2013, the Company owed two directors a total of $60,350 and $57,237, respectively, related to expenses incurred on behalf of the Company related to corporate startup costs and intellectual property.
Going Concern Qualification
The Company has incurred significant losses and cash used in operations, and such losses and use of cash are expected to continue. The Company’s Independent Registered Public Accounting Firm has included a "Going Concern Qualification" in their report for the years ended June 30, 2014 and 2013. In addition, the Company has negative working capital. The foregoing raises substantial doubt about the Company's ability to continue as a going concern. Management's plans include seeking additional capital or debt financing. There is no guarantee that additional capital or debt financing will be available when and to the extent required, or that if available, it will be on terms acceptable to the Company. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty. The "Going Concern Qualification" might make it substantially more difficult to raise capital.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources.
Cautionary Note Regarding Forward-Looking Statements
This report includes forward-looking statements regarding our liquidity, our ability to complete financing, our ability to purchase capital expenditures, expected proceeds, growth of our business including entering into future agreements with companies, and plans to successfully develop and obtain approval to market our product.
All statements other than statements of historical facts contained in this report, including statements regarding our future financial position, liquidity, business strategy and plans and objectives of management for future operations, are forward-looking statements. The words “believe,” “may,” “estimate,” “continue,” “anticipate,” “intend,” “should,” “plan,” “could,” “target,” “potential,” “is likely,” “will,” “expect” and similar expressions, as they relate to us, are intended to identify forward-looking statements. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our financial condition, results of operations, business strategy and financial needs.
The results anticipated by any or all of these forward-looking statements might not occur. Important factors, uncertainties and risks that may cause actual results to differ materially from these forward-looking statements are contained in the risk factors that follow. We undertake no obligation to publicly update or revise any forward-looking statements, whether as the result of new information, future events or otherwise. For more information regarding some of the ongoing risks and uncertainties of our business, see the risk factors which follow.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk.
Not applicable to smaller reporting companies.
| 37 |
Item 8. Financial Statements and Supplementary Data.
INDEX TO FINANCIAL STATEMENTS
| F-1 |

Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of:
Propanc Health Group Corporation
We have audited the accompanying consolidated balance sheets of Propanc Health Group Corporation and Subsidiary (a development stage company) at June 30, 2014 and 2013 and the related consolidated statements of operations and comprehensive loss, changes in stockholders’ deficit and cash flows for each of the two years in the period ended June 30, 2014. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall consolidated financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of Propanc Health Group Corporation and Subsidiary at June 30, 2014 and 2013 and the consolidated results of its operations and its cash flows for each of the two years in the period ended June 30, 2014, in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the consolidated financial statements, the Company had no revenues and had a net loss of $829,564 and net cash used in operations of $226,442. Additionally, as of June 30, 2014, the Company had a working capital deficit, stockholders’ deficit, and accumulated deficit of $1,408,314, $1,408,314, and $17,552,917. These matters raise substantial doubt about the Company’s ability to continue as a going concern. Management’s Plan in regards to these matters is also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/S/ Salberg & Company, P.A.
SALBERG & COMPANY, P.A.
Boca Raton, Florida
October 14, 2014

| F-2 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
CONSOLIDATED BALANCE SHEETS
| June 30, 2014 | June 30, 2013 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS: | ||||||||
| Cash | $ | 87,799 | $ | - | ||||
| GST tax receivable | 946 | 1,209 | ||||||
| Prepaid expenses and other current assets | 25,000 | - | ||||||
| TOTAL CURRENT ASSETS | 113,745 | 1,209 | ||||||
| Property and Equipment, net | - | 538 | ||||||
| TOTAL ASSETS | $ | 113,745 | $ | 1,747 | ||||
| LIABILITIES AND STOCKHOLDERS' DEFICIT | ||||||||
| CURRENT LIABILITIES: | ||||||||
| Bank overdraft | $ | - | $ | 5 | ||||
| Accounts payable | 350,004 | 272,596 | ||||||
| Accrued expenses and other payables | 422,326 | 251,439 | ||||||
| Other loans | 33,909 | 32,879 | ||||||
| Convertible notes, net | 272,424 | - | ||||||
| Warrant derivative liability | 158,244 | - | ||||||
| Due to directors - related parties | 60,350 | 57,237 | ||||||
| Loans from directors and officer - related parties | 161,975 | 130,689 | ||||||
| Employee benefit liability | 62,827 | 49,378 | ||||||
| TOTAL CURRENT LIABILITIES | 1,522,059 | 794,223 | ||||||
| Commitments and Contingencies (See Note 9) | ||||||||
| STOCKHOLDERS' DEFICIT: | ||||||||
| Preferred stock, $0.01 par value; 10,000,000 shares authorized; zero shares issued and outstanding as of June 30, 2014 and June 30, 2013, respectively | - | - | ||||||
| Common stock, $0.001 par value; 100,000,000 shares authorized; 72,684,767 and 70,632,267 shares issued and outstanding as of June 30, 2014 and June 30, 2013, respectively | 72,685 | 70,632 | ||||||
| Common stock issuable, $0.001 par value; 0 and 25,000 shares issued and outstanding as of June 30, 2014 and June 30, 2013, respectively | - | 25 | ||||||
| Additional paid-in capital | 16,374,781 | 16,104,809 | ||||||
| Accumulated other comprehensive loss | (302,863 | ) | (244,589 | ) | ||||
| Accumulated deficit | (17,552,917 | ) | (16,723,353 | ) | ||||
| TOTAL STOCKHOLDERS' DEFICIT | (1,408,314 | ) | (792,476 | ) | ||||
| TOTAL LIABILITIES AND STOCKHOLDERS' DEFICIT | $ | 113,745 | $ | 1,747 | ||||
The accompanying notes are an integral part of these consolidated financial statements
| F-3 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
FOR YEARS ENDED JUNE 30, 2014 AND 2013
| Year Ended June 30, | ||||||||
| 2014 | 2013 | |||||||
| REVENUE | ||||||||
| Royalty revenue - related party | $ | - | $ | - | ||||
| OPERATING EXPENSES | ||||||||
| Administration expenses | 742,037 | 1,336,482 | ||||||
| Occupancy expenses | 11,016 | 13,074 | ||||||
| Research and development | 8,168 | 12,344 | ||||||
| TOTAL OPERATING EXPENSES | 761,221 | 1,361,900 | ||||||
| LOSS FROM OPERATIONS | (761,221 | ) | (1,361,900 | ) | ||||
| OTHER INCOME (EXPENSE) | ||||||||
| Interest expense | (93,147 | ) | (5,465 | ) | ||||
| Interest income | 18 | - | ||||||
| Change in fair value of warrant derivative liability | (16,522 | ) | - | |||||
| Loss on debt settlements, net | - | (108,185 | ) | |||||
| Foreign currency transaction loss | (6,959 | ) | (27,549 | ) | ||||
| TOTAL OTHER INCOME (EXPENSE) | (116,610 | ) | (141,199 | ) | ||||
| LOSS BEFORE INCOME TAXES | (877,831 | ) | (1,503,099 | ) | ||||
| INCOME TAX BENEFIT | 48,267 | 60,461 | ||||||
| NET LOSS | (829,564 | ) | (1,442,638 | ) | ||||
| OTHER COMPREHENSIVE (LOSS) GAIN | ||||||||
| Foreign currency translation (loss) gain | (58,274 | ) | 114,328 | |||||
| COMPREHENSIVE LOSS | $ | (887,838 | ) | $ | (1,328,310 | ) | ||
| BASIC AND DILUTED NET LOSS PER SHARE | $ | (0.01 | ) | $ | (0.02 | ) | ||
| BASIC AND DILUTED WEIGHTED AVERAGE SHARES OUTSTANDING | 72,350,555 | 72,365,530 | ||||||
The accompanying notes are an integral part of these consolidated financial statements
| F-4 |
PROPANC HEALTH GROUP AND SUBSIDIARY
CONSOLIDATED STATEMENT OF CHANGES IN STOCKHOLDERS' DEFICIT
FOR THE YEARS ENDED JUNE 30, 2014 AND 2013
| Accumulated | ||||||||||||||||||||||||||||||||||||
| Other | Total | |||||||||||||||||||||||||||||||||||
| Common Stock Issuable | Common Stock | Subscription | Additional | Comprehensive | Stockholders' | |||||||||||||||||||||||||||||||
| No. of Shares | Value | No. of Shares | Value | Receivable | Paid-in Capital | Accumulated Deficit | loss | Deficit | ||||||||||||||||||||||||||||
| Balance at June 30, 2012 | 5,877 | $ | 6 | 72,705,569 | $ | 72,706 | $ | (325 | ) | $ | 15,029,326 | $ | (15,280,715 | ) | $ | (358,917 | ) | $ | (537,919 | ) | ||||||||||||||||
| Issuance of common stock for conversion of convertible debt | - | - | 51,264 | 51 | - | 76,845 | - | - | 76,896 | |||||||||||||||||||||||||||
| Issuance of common stock for services | (5,877 | ) | (6 | ) | 5,877 | 6 | - | - | - | - | - | |||||||||||||||||||||||||
| Write off of subscription receivable | - | - | - | - | 325 | - | - | - | 325 | |||||||||||||||||||||||||||
| Consultant stock expense | - | - | - | - | - | 48,000 | - | - | 48,000 | |||||||||||||||||||||||||||
| Shares issued under voluntary ratchet | - | - | 214,089 | 214 | - | 138,944 | - | - | 139,158 | |||||||||||||||||||||||||||
| Issuance of common stock for conversion of accrued expenses | - | - | 225,000 | 225 | - | 146,025 | - | - | 146,250 | |||||||||||||||||||||||||||
| Issuance of stock for services | 25,000 | 25 | 1,012,500 | 1,013 | - | 662,087 | - | - | 663,125 | |||||||||||||||||||||||||||
| Cancellation of shares | - | - | (3,582,032 | ) | (3,582 | ) | - | 3,582 | - | - | - | |||||||||||||||||||||||||
| Foreign currency translation gain | - | - | - | - | - | - | - | 114,328 | 114,328 | |||||||||||||||||||||||||||
| Net loss, 2013 | - | - | - | - | - | - | (1,442,638 | ) | - | (1,442,638 | ) | |||||||||||||||||||||||||
| Balance at June 30, 2013 | 25,000 | $ | 25 | 70,632,267 | $ | 70,632 | $ | - | $ | 16,104,809 | $ | (16,723,353 | ) | $ | (244,589 | ) | $ | (792,476 | ) | |||||||||||||||||
| Issuance of stock for services | (25,000 | ) | (25 | ) | 1,915,000 | 1,915 | - | 242,610 | - | - | 244,500 | |||||||||||||||||||||||||
| Issuance of common stock for conversion of accrued expenses | - | - | 137,500 | 138 | - | 27,362 | - | - | 27,500 | |||||||||||||||||||||||||||
| Foreign currency translation loss | - | - | - | - | - | - | - | (58,274 | ) | (58,274 | ) | |||||||||||||||||||||||||
| Net loss, 2014 | - | - | - | - | - | - | (829,564 | ) | - | (829,564 | ) | |||||||||||||||||||||||||
| Balance at June 30, 2014 | - | $ | - | 72,684,767 | $ | 72,685 | $ | - | $ | 16,374,781 | $ | (17,552,917 | ) | $ | (302,863 | ) | $ | (1,408,314 | ) | |||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements
| F-5 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CASH FLOWS
FOR YEARS ENDED JUNE 30, 2014 AND 2013
| Year Ended June 30, | ||||||||
| 2014 | 2013 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss | $ | (829,564 | ) | $ | (1,442,638 | ) | ||
| Adjustments to Reconcile Net loss to Net Cash Used in Operating Activities: | ||||||||
| Issuance of common stock for services and voluntary ratchet | 237,833 | 803,324 | ||||||
| Loss on settlement | - | 108,185 | ||||||
| Consultant stock expense | - | 48,000 | ||||||
| Amortization of prepaid shares issued for services | 6,667 | 38,937 | ||||||
| Write off of subscription receivable | - | 325 | ||||||
| Foreign currency transaction (loss) gain | - | 27,549 | ||||||
| Depreciation expense | 538 | 4,062 | ||||||
| Amortization of debt discount | 49,029 | - | ||||||
| Change in fair value of warrant derivative liability | 16,522 | - | ||||||
| Accretion of put premium | 27,187 | - | ||||||
| Changes in Assets and Liabilities: | ||||||||
| Escrow account | - | 333 | ||||||
| Prepaid expenses and other assets | 294 | 1,293 | ||||||
| Accounts payable | 94,607 | 121,649 | ||||||
| Employee benefit liability | 11,598 | 12,965 | ||||||
| Accrued expenses | 158,847 | 147,369 | ||||||
| NET CASH USED IN OPERATING ACTIVITIES | (226,442 | ) | (128,647 | ) | ||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Bank overdraft | (6 | ) | 5 | |||||
| Loan repayments to principal stockholder | (45,512 | ) | (32,860 | ) | ||||
| Proceeds from convertible promissory notes, net of costs | 273,959 | - | ||||||
| Loan proceeds | 10,542 | 37,506 | ||||||
| Loan proceeds from principal stockholder | 72,158 | 123,814 | ||||||
| NET CASH PROVIDED BY FINANCING ACTIVITIES | 311,141 | 128,465 | ||||||
| Effect of exchange rate changes on cash | 3,100 | 3 | ||||||
| NET INCREASE (DECREASE) IN CASH | 87,799 | (179 | ) | |||||
| CASH AT BEGINNING OF YEAR | - | 179 | ||||||
| CASH AT END OF YEAR | $ | 87,799 | $ | - | ||||
| Supplemental Disclosure of Cash Flow Information | ||||||||
| Cash paid during the period: | ||||||||
| Interest | $ | - | $ | - | ||||
| Income Tax | $ | - | $ | - | ||||
| Supplemental Disclosure of Non-Cash Investing and Financing Activities | ||||||||
| Conversion of accrued interest to common stock | $ | - | $ | 1,896 | ||||
| Prepaid common stock issued for services | $ | 6,667 | $ | - | ||||
| Conversion of accrued expenses to common stock | $ | 27,500 | $ | 37,500 | ||||
| Conversion of convertible notes and accrued interest to common stock | $ | - | $ | 75,000 | ||||
| Discounts related to warrants issued with convertible debenture | $ | 133,095 | $ | - | ||||
| Discounts related to lender costs | $ | 30,000 | $ | - | ||||
| Conversion of loan payable to convertible debenture | $ | 27,963 | $ | - | ||||
| Prepaid settlement fee paid through issuance of convertible note | $ | 25,000 | $ | - | ||||
The accompanying notes are an integral part of these consolidated financial statements
| F-6 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
NOTE 1 – NATURE OF OPERATIONS AND SUMMARY OF SIGNIFICANT ACCOUNTING AND REPORTING POLICIES
Nature of Operations
Propanc Health Group Corporation, formerly Propanc PTY LTD, ("the Company", "we", "us", "our") is a development stage enterprise. Propanc PTY LTD was incorporated in Melbourne, Victoria Australia on October 15, 2007, and is based in Richmond, Victoria Australia. Since inception, substantially all of the efforts of the Company have been the development of new cancer treatments targeting high risk patients who need a follow up, nontoxic, long term therapy which prevents the cancer from returning and spreading. The Company anticipates establishing global markets for its technologies.
On November 23, 2010, Propanc Health Group Corporation was incorporated in the state of Delaware. In January 2011, to reorganize the Company, Propanc Health Group Corporation acquired all of the outstanding shares of Propanc PTY LTD on a one-for-one basis making it a wholly-owned subsidiary.
Principals of Consolidation
The consolidated financial statements include the accounts of Propanc Health Group Corporation and its wholly-owned subsidiary, Propanc PTY LTD. All significant inter-company balances and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from these estimates. Significant estimates in the accompanying consolidated financial statements include the estimates of valuation of derivatives, valuation of beneficial conversion features on convertible debt, allowance for uncollectable receivables, valuation of equity based instruments issued for other than cash, the valuation allowance on deferred tax assets and foreign currency translation due to certain average exchange rates applied in lieu of spot rates on translation dates.
Foreign Currency Translation and Comprehensive Income (Loss)
The Company’s functional currency is the Australian dollar (AUD). For financial reporting purposes, the Australian dollar has been translated into United States dollars ($) and/or USD as the reporting currency. Assets and liabilities are translated at the exchange rate in effect at the balance sheet date. Revenues and expenses are translated at the average rate of exchange prevailing during the reporting period. Equity transactions are translated at each historical transaction date spot rate. Translation adjustments arising from the use of different exchange rates from period to period are included as a component of stockholders’ equity (deficit) as “accumulated other comprehensive income (loss).” Gains and losses resulting from foreign currency transactions are included in the statement of operations and comprehensive loss as other income (expense). There have been no significant fluctuations in the exchange rate for the conversion of Australian dollars to USD after the balance sheet date.
Comprehensive loss for all periods presented, includes only foreign currency translation gains (losses).
| F-7 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
Changes in Accumulated Other Comprehensive Income (Loss) by Component during 2013 and 2014 was as follows:
| Foreign Currency Items: | ||||
| Beginning balance June 30, 2012 | $ | (358,917 | ) | |
| Other comprehensive income before reclassification | 114,328 | |||
| Balance, June 30, 2013 | $ | (244,589 | ) | |
| Other comprehensive loss before reclassification | (58,274 | ) | ||
| Amounts reclassified from accumulated other comprehensive income (loss) | - | |||
| Ending balance, June 30, 2014 | $ | (302,863 | ) | |
Fair Value of Financial Instruments and Fair Value Measurements
We measure our financial assets and liabilities in accordance with United States generally accepted accounting principles. For certain of our financial instruments, including cash and cash equivalents, accounts and other receivables, accounts payable and accrued and other liabilities, the carrying amounts approximate fair value due to their short maturities. Amounts recorded for loans payable, also approximate fair value because current interest rates available to us for debt with similar terms and maturities are substantially the same.
We adopted accounting guidance for fair value measurements of financial assets and liabilities. The adoption did not have a material impact on our results of operations, financial position or liquidity. This standard defines fair value, provides guidance for measuring fair value and requires certain disclosures. This standard does not require any new fair value measurements, but rather applies to all other accounting pronouncements that require or permit fair value measurements. This guidance does not apply to measurements related to share-based payments. This guidance discusses valuation techniques, such as the market approach (comparable market prices), the income approach (present value of future income or cash flow), and the cost approach (cost to replace the service capacity of an asset or replacement cost). The guidance utilizes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three broad levels. The following is a brief description of those three levels:
Level 1: Observable inputs such as quoted prices (unadjusted) in active markets for identical assets or liabilities.
Level 2: Inputs other than quoted prices that are observable, either directly or indirectly. These include quoted prices for similar assets or liabilities in active markets and quoted prices for identical or similar assets or liabilities in markets that are not active.
Level 3: Unobservable inputs in which little or no market data exists, therefore developed using estimates and assumptions developed by us, which reflect those that a market participant would use.
Derivative Instruments
ASC Topic 815, Derivatives and Hedging (“ASC Topic 815”), establishes accounting and reporting standards for derivative instruments and for hedging activities by requiring that all derivatives be recognized in the balance sheet and measured at fair value. Gains or losses resulting from changes in the fair value of derivatives are recognized in earnings or recorded in other comprehensive income (loss) depending on the purpose of the derivatives and whether they qualify and have been designated for hedge accounting treatment. The Company does not have any derivative instruments for which it has applied hedge accounting treatment.
| F-8 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
Cash and Cash Equivalents
Cash and cash equivalents include cash on hand and at banks, short-term deposits with an original maturity of three months or less held at call with financial institutions, and bank overdrafts. Bank overdrafts are reflected as a current liability on the balance sheets. There were no cash equivalents as of June 30, 2014 or 2013.
Receivables
As amounts become uncollectible, they will be charged to an allowance or operations in the period when a determination of uncollectability is made. Any estimates of potentially uncollectible customer accounts receivable will be made based on an analysis of individual customer and historical write-off experience. The Company’s analysis included the age of the receivable account, creditworthiness, and general economic conditions.
Property, Plant, and Equipment
Property and equipment are stated at cost, net of accumulated depreciation. Expenditures for maintenance and repairs are expensed as incurred; additions, renewals, and betterments are capitalized. When property and equipment are retired or otherwise disposed of, the related cost and accumulated depreciation are removed from the respective accounts, and any gain or loss is included in operations. Depreciation of property and equipment is provided using the declining balance method. The depreciable amount is the cost less its residual value.
The estimated useful lives are as follows:
Machinery and equipment 3 years
Patents
Patent costs are stated at cost and reclassified to intangible assets and amortized on a straight-line basis over the estimated future periods if and once the patent has been granted by a regulatory agency, however, the Company will expense any costs as long as the Company is in the development stage. Accordingly, as the Company's product is not currently approved for market, during 2012, the Company wrote-off approximately $27,000 of previously capitalized patent costs related to various applications. Any patent costs incurred in 2013 and 2014 were expensed immediately. Currently, the Company has one International patent pending which was jointly applied for by the company and another entity.
The Company received grant status in South Africa and more recently in New Zealand. In addition, the United States Patent and Trademark Office or USPTO and European Patent Office or EPO have made preliminary indications that key features of our technology are patentable. The Company is presently working towards securing a patent in each region, covering as many aspects of its technology as possible, whilst also actively seeking protection throughout Eastern Europe, Asia and South America.
Individual countries and regions, include United States, Canada, Japan, Brazil, China, Mexico, Hong Kong, Singapore, Israel, Chile, Peru, Malaysia, Vietnam, Indonesia, Europe, Russia, India, Australia and South Korea. The patent is granted in South Africa and New Zealand.
Impairment of Long-Lived Assets
In accordance with ASC 360-10, Long-lived assets, which include property and equipment and intangible assets, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of long-lived assets to be held and used is measured by a comparison of the carrying amount of an asset to the estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated undiscounted future cash flows, an impairment charge is recognized by the amount by which the carrying amount of the asset exceeds the fair value of the assets. Fair value is generally determined using the asset’s expected future discounted cash flows or market value, if readily determinable.
| F-9 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
Employee Benefit/Liability
Liabilities arising in respect of wages and salaries, annual leave, accumulated sick leave and any other employee benefits expected to be settled within twelve months of the reporting date are measured at their nominal amounts based on remuneration rates which are expected to be paid when the liability is settled. All other employee benefit liabilities are measured at the present value of the estimated future cash outflow to be made in respect of services provided by employees up to the reporting date. All employee liabilities are owed within the next twelve months.
Australian Goods and Services Tax (GST)
Revenues, expenses and balance sheet items are recognized net of the amount of GST except payable and receivable balances which are shown inclusive of GST. The GST incurred is payable on revenues to, and recoverable on purchases from, the Australian Taxation Office.
Cash flows are presented in the statements of cash flow on a gross basis, except for the GST component of investing and financing activities, which are disclosed as operating cash flows.
As of June 30, 2014 and 2013 the Company was owed $946 and $1,209 from the Australian Taxation Office. These amounts were fully collected subsequent to the balance sheet reporting dates.
Income Taxes
The Company is governed by Australia and United States income tax laws, which are administered by the Australian Taxation Office and the United States Internal Revenue Service, respectively. The Company follows FASB ASC 740 when accounting for income taxes, which requires an asset and liability approach to financial accounting and reporting for income taxes. Deferred income tax assets and liabilities are computed annually for temporary differences between the financial statements and tax bases of assets and liabilities that will result in taxable or deductible amounts in the future based on enacted tax laws and rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized. Income tax expense is the tax payable or refundable for the period plus or minus the change during the period in deferred tax assets and liabilities.
The Company adopted provisions of ASC 740, Sections 25 through 60, “Accounting for Uncertainty in Income Taxes." These sections provide detailed guidance for the financial statement recognition, measurement and disclosure of uncertain tax positions recognized in the financial statements. Tax positions must meet a “more-likely-than-not” recognition threshold at the effective date to be recognized upon the adoption of ASC 740 and in subsequent periods. Upon the adoption of ASC 740, the Company had no unrecognized tax benefits. During the years ended June 30, 2014 and 2013 no adjustments were recognized for uncertain tax benefits. The years 2008 through 2014 are subject to examination by the Australian Taxation Office. The years ended June 30, 2011 through 2014 is subject to examination by the United States Internal Revenue Service.
Research and Development Tax Credits
The Company may apply for Research and Development tax concessions with the Australian Taxation Office on an annual basis. Although the amount is possible to estimate at year end, the Australian Taxation Office may reject or materially alter the claim amount. Accordingly, the Company does not recognize the benefit of the claim amount until cash receipt since collectability is not certain until such time. The tax concession is a refundable credit. If the Company has net income then the Company can receive the credit which reduces its income tax liability. If the Company has net losses then the Company may still receive a cash payment for the credit, however, the Company's net operating loss carryforwards are reduced by the gross equivalent loss that would produce the credit amount when the income tax rate is applied to that gross amount. The concession is recognized as an income tax benefit, in operations, upon receipt.
| F-10 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
During the years-ended ended June 30, 2014 and 2013, the Company applied for and received from the Australian Taxation Office a research and development tax credit in the amount of $48,267 and $60,461 respectively, which is reflected as an income tax benefit in the accompanying consolidated statement of operations and comprehensive loss.
Stock Based Compensation
The Company records stock based compensation in accordance with ASC section 718, “Stock Compensation” and Staff Accounting Bulletin (SAB) No. 107 (SAB 107) issued by the Securities and Exchange Commission (SEC) in March 2005 regarding its interpretation of ASC 718. ASC 718 requires the fair value of all stock-based employee compensation awarded to employees to be recorded as an expense over the related requisite service period. The statement also requires the recognition of compensation expense for the fair value of any unvested stock option awards outstanding at the date of adoption. The Company values any employee or non-employee stock based compensation at fair value using the Black-Scholes Option Pricing Model.
The Company accounts for non-employee share-based awards in accordance with the measurement and recognition criteria of ASC 505-50 “Equity-Based Payments to Non-Employees”.
Revenue Recognition
In accordance with Securities and Exchange Commission (SEC) Staff Accounting Bulletin (SAB) No. 104, Revenue Recognition, (codified in ASC 605) the Company recognizes revenue when (i) persuasive evidence of a customer or distributor arrangement exists or acceptance occurs, (ii) a retailer, distributor or wholesaler receives the goods, (iii) the price is fixed or determinable, and (iv) collectability of the sales revenues is reasonably assured. Subject to these criteria, the Company recognizes revenue relating to royalties on product sales in the period in which the sale occurs and the royalty term has begun.
Start-up Costs
In accordance with ASC 720-15-15, start-up costs are expensed as incurred.
Research and Development Costs
In accordance with ASC 730-10, Research and development costs are expensed when incurred. Total research and development costs for the years ended June 30, 2014 and 2013 were $8,168 and $12,344, respectively.
Basic and Diluted Net Loss Per Common Share
Basic net loss per share is computed by dividing the net loss by the weighted average number of common shares outstanding during the period. Diluted net loss per common share is computed by dividing the net loss by the weighted average number of common shares outstanding for the period and, if dilutive, potential common shares outstanding during the period. Potentially dilutive securities consist of the incremental common shares issuable upon exercise of common stock equivalents such as stock options and convertible debt instruments. Potentially dilutive securities are excluded from the computation if their effect is anti-dilutive. As a result, the basic and diluted per share amounts for all periods presented are identical. As of June 30, 2013, there were no potentially dilutive securities. As of June 30, 2014, there were 3,000,000 warrants outstanding and four convertible notes payable that are convertible into 6,069,667 common shares which are considered dilutive securities which were excluded from the computation since the effect is anti-dilutive.
| F-11 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
Recently Adopted Accounting Pronouncements
Financial Accounting Standards Board, Accounting Standard Updates which are not effective until after June 30, 2014 are not expected to have a significant effect on the Company’s unaudited consolidated financial position or results of operations. The Company implemented the following at June 30, 2014:
In June 2014, the FASB issued ASU No. 2014-10, which amended Accounting Standards Codification (ASC) Topic 915 Development Stage Entities. The amendment eliminates certain financial reporting requirements surrounding development stage entities, including an amendment to the variable interest entities guidance in ASC Topic 810, Consolidation. The amendment removes the definition of a development stage entity from the ASC, thereby removing the financial reporting distinction between development stage entities and other entities from U.S. GAAP. Consequently, the amendment eliminates the requirements for development stage entities to (1) present inception-to-date information in the statements of income, cash flows and shareholder equity, (2) label the financial statements as those of a development stage entity, (3) disclose a description of the development stage activities in which the entity is engaged, and (4) disclose the first year in which the entity is no longer a development stage entity that in prior years it had been in the development stage.
This amendment is effective for fiscal years beginning after December 15, 2014, and interim periods therein. Early application of each of the amendments is permitted for any annual reporting period or interim period for which the entity's financial statements have not yet been issued. The Company has made the election to early adopt this amendment effective June 30, 2014 and, as a result, the Company is no longer presenting or disclosing the information previously required under Topic 915. The early adoption was made to reduce data maintenance by removing all incremental financial reporting requirements for development stage entities. The adoption of this amendment alters the disclosure requirements of the Company, but it does not have any material impact on the Company's financial position or results of operations for the current or any prior reporting periods.
NOTE 2 – GOING CONCERN
The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America, which contemplate continuation of the Company as a going concern. For the year ended June 30, 2014, the Company had no revenues and had a net loss of $829,564 and net cash used in operations of $226,442. Additionally, as of June 30, 2014, the company had a working capital deficit, stockholders' deficit and accumulated deficit of $1,408,314, $1,408,314, and $17,552,917 respectively. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. The consolidated financial statements do not include any adjustments to reflect the possible future effect on the recoverability and classification of assets or the amounts and classifications of liabilities that may result from the outcome of this uncertainty.
Successful completion of the Company’s development program and, ultimately, the attainment of profitable operations are dependent upon future events, including obtaining adequate financing to fulfill its development activities, acceptance of the Company's International patent application and achieving a level of sales adequate to support the Company’s cost structure. However, there can be no assurances that the Company will be able to secure additional equity investment or achieve an adequate sales level.
| F-12 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
NOTE 3 – PROPERTY AND EQUIPMENT
Property, plant, and equipment consist of the following as of June 30,
| 2014 | 2013 | |||||||
| Office equipment at cost | $ | 14,968 | $ | 14,513 | ||||
| Less: Accumulated depreciation | (14,968 | ) | (13,975 | ) | ||||
| Total property, plant, and equipment | $ | - | $ | 538 | ||||
Depreciation expense for the years ended June 30, 2014 and 2013 were $538 and $4,062, respectively.
NOTE 4 – DUE TO DIRECTORS - RELATED PARTIES
Due to directors - related parties represents unsecured advances made by the directors for operating expenses on behalf of the Company such as intellectual property and formation expenses. The expenses were paid for on behalf of the Company are due upon demand. The Company is currently not being charged interest under these advances. The total amount owed these directors at June 30, 2014 and 2013 is $60,350 and $57,237 respectively.
NOTE 5 – LOANS
Loans from Directors - Related Parties
Loans from Directors and Officer at June 30, 2014 and 2013 were $161,975 and $130,689, respectively. The loans bear interest at rates of prime + 2% (5.25% at June 30, 2014) and are all past their due date and in default.
Other Loans from Unrelated Parties
Loans from two unrelated parties were received during 2013 totaling $33,614. They bear interest at 10% and as of June 30, 2014 one was past its due date and in default and the other, with a September 30, 2013 balance of $27,963 was exchanged for a convertible debenture as discussed below in Note 6. Accrued interest was $994 at June 30, 2014.
A loan from an unrelated party was received during the year ended June 30, 2014 totaling $9,419. It bears interest at 10%. Accrued interest was $425 at June 30, 2014.
A Loan from an unrelated party was received during the year-ended June 30, 2014 totaling $18,839 and is non-interest bearing.
Accordingly, other loans totaled $33,909 at June 30, 2014.
NOTE 6 – CONVERTIBLE NOTES
Convertible notes at June 30, 2014 were as follows:
| Convertible notes and debenture | $ | 366,296 | ||
| Unamortized discounts | (121,059 | ) | ||
| Premium | 27,187 | |||
| Convertible notes, net | $ | 272,424 |
| F-13 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
On September 30, 2013 the Company’s subsidiary issued a Debenture for $139,680 (AUD$150,000) plus warrants for 3,000,000 common shares of the Company. The Company agreed to pay 12% interest on the principal amount and the maturity date is December 31, 2015. This debenture rolls into it $27,963 of loans outstanding at June 30, 2013 (see Note 5), an August 2013 note of $63,196 along with September advances of $46,446 and accrued interest. The debenture is convertible only at the Company’s option into common stock at $0.0698 per share and is convertible at that same rate by the lender only upon default by the Company, as defined in the debenture. The warrants were determined to be derivative instruments due to the variable exercise price of the warrants which is initially $0.0698 and subject to adjustment if the Company issues shares at a price below the initial exercise price. Accordingly, the fair value of the warrants was determined using a Black-Scholes option pricing model with a stock price of $0.20, exercise price of $0.0698, volatility of 53% based on the comparative companies method since the Company’s stock is very thinly traded, an expected term of 27 months based on the debenture term and a risk free rate of 0.4%. The approximate initial $400,000 value of the warrants was recorded as a derivative liability in the accompanying consolidated balance sheet, along with a debt discount of approximately $140,000 and change in warrant derivative liability of approximately $260,000 as an expense for the three months ended September 30, 2013. (See note 12 for current period re-measurement)
In May 2014, the Company issued a 10% convertible promissory note for $25,000 as a prepaid fee for services to be provided under a settlement and stipulation agreement as discussed in Note 13. The note and all accrued interest is due on November 8, 2014. The note is convertible immediately at 50% of the lowest closing bid price in the 30 days prior to conversion.
On May 29, 2014, the Company issued a convertible note payable for $75,000. The Company agreed to pay 8% interest per annum on the principal amount and the maturity date is May 29, 2015. The note is convertible at the option of the holder at any time after 180 days at a rate of 55% of the lowest trading bid price of the Company’s common stock for the ten prior trading including the date upon which the conversion notice was received. The convertible note is treated as stock settled debt under ASC 480 and accordingly the Company is accreting a $61,364 put premium over 180 days from the execution of the convertible note. Through June 30, 2014, the Company has accreted $10,275 of the put premium. Accrued interest as of June 30, 2014 was $526.
On May 29, 2014, the Company issued a second convertible note payable for $75,000. The Company agreed to pay 8% interest per annum on the principal amount and the maturity date is May 29, 2015. The note is convertible at the option of the holder at any time after 180 days at a rate of 55% of the lowest trading bid price of the Company’s common stock for the ten prior trading including the date upon which the conversion notice was received. The convertible note is treated as stock settled debt under ASC 480 and accordingly the Company is accreting a $61,364 put premium over 180 days from the execution of the convertible note. Through June 30, 2014, the Company has accreted $10,275 of the put premium. Accrued interest as of June 30, 2014 was $526.
On May 30, 2014, the Company issued a third convertible note payable for $50,000. The Company agreed to pay 8% interest per annum on the principal amount and the maturity date is May 29, 2015. The note is convertible at the option of the holder at any time after 180 days at a rate of 55% of the lowest trading bid price of the Company’s common stock for the ten prior trading including the date upon which the conversion notice was received. The convertible note is treated as stock settled debt under ASC 480 and accordingly the Company is accreting a $40,909 put premium over 180 days from the execution of the convertible note. Through June 30, 2014, the Company has accreted $6,636 of the put premium. Accrued interest as of June 30, 2014 was $340.
The Company recorded $30,000 of debt discounts for fees paid to lenders related to the above note issuances. Amortization of the discounts through June 30, 2014 was $3,133.
In addition to each of the above initial convertible promissory notes (“initial convertible notes”), the Company issued to each lender another convertible promissory note for the same amounts of $75,000, $75,000 and $50,000 termed "back-end notes". These notes have the same terms as the initial notes. Each back-end note shall initially be paid for by an offsetting promissory note issued to the Company by the lender ("Note receivable") provided that prior to the conversion of the Back-End Notes, the holders must have paid off the Notes receivable in cash. The Notes receivable are due on January 30, 2015, unless the Company does not meet the “current public information” requirement pursuant to Rule 144, in which case both the Back-End Notes and the Notes receivable may be both cancelled. The Notes receivable are initially secured by the pledge of the back end Notes, but may be exchanged for other collateral with an appraised value of at least $50,000, upon Company’s approval following a three (3) day written notice to the Company. The term of the Notes receivable and the Back-End Notes are one year, upon which the outstanding principal and interest is payable. The amount funded plus accrued interest under Back-End Notes are convertible into Common Stock at any time after the requisite Rule 144 holding period (subject to the condition above for the Back-End Notes), at a conversion price equal to 55% of the lowest trading bid price in the ten (10) trading days prior to the conversion.
| F-14 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
In the event the Company redeems the initial convertible promissory notes in full, the Company is required to pay off all principal, interest and any other amounts owing multiplied by i) 130% if prepaid within 60 days of the issuance date; ii) 140% if prepaid 60 but less than 121 days after the issuance date; and (iii) 150% if prepaid 120 but less than 180 days after the issuance date. There shall be no redemption after the 180th day. The Back-End Notes may not be prepaid, except that if the initial Convertible Notes are redeemed by the Company within six months of their issuance, all obligations of the Company and holders under the Back-End Note and the Notes receivable will be deemed satisfied and such notes shall automatically be deemed cancelled and of no further force or effect.
In the event of two specific defaults, which include the maintenance of a minimum trading price and an aggregate dollar trading volume of the Company's common shares, the holders may cancel the back-end notes and the related notes receivable and otherwise in the event of other defaults as defined in the securities purchase agreement, the amount of principal and accrued interest will become immediately due and payable and may be offset by amounts due to the Company by the holders. Additionally, the back-end notes will bear default interest at a rate of 16% per annum, or the highest rate of interest permitted by law.
Since the back-end notes are not convertible until the notes receivable are paid and also not for 180 days from the note dates, and the notes receivable and notes payable have a right a setoff, the notes receivable and back-end notes and related accrued interest receivable and payable have been netted for presentation purposes on the accompanying consolidated balance sheet.
NOTE 7 – INCOME TAXES
The Company follows ASC 740-10-10, under which an entity recognizes deferred tax assets and liabilities for future tax consequences or for events that were previously recognized in the Company’s financial statements or tax returns. The measurement of deferred tax assets and liabilities is based on enacted tax law provisions. The effects of future changes in tax laws or rates are not anticipated. As of June 30, 2014, the Company operated exclusively in Australia. The Company was wholly subject to Australia income tax laws and regulations, which are administered by the Australian Taxation Office for the years ended June 30, 2014 and 2013 and all prior years.
On November 23, 2010, Propanc Health Group Corporation was incorporated in the state of Delaware. In January 2011, Propanc Health Group Corporation acquired all of the outstanding shares of Propanc PTY LTD on a one-for-one basis making it a wholly-owned subsidiary. As a result of these transactions, the Company is subject to the income tax laws of both the United States and Australia for the years ended June 30, 2013 and 2014. For the years ended June 30, 2014 and 2013, all the Company’s loss before income taxes resulted entirely from its Australian activities and its taxable loss was only subject to Australian tax law.
At June 30, 2014, the Company has a net operating loss (NOL) for Australian tax purposes only, that approximates $10,586,000. Consequently, the Company may have NOL carryforwards available for income tax purposes, which will continue to be available until they are recovered through earning taxable income. Deferred tax assets would arise from the recognition of anticipated utilization of these net operating losses to offset future taxable income. The NOL is subject to a reduction of $1,399,918 for research and development credits granted by the Australian Taxation Office through June 30, 2014.
| F-15 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
The components for the provision for income taxes are as follows:
| Year Ended | ||||||||
| June 30, | June 30, | |||||||
| 2014 | 2013 | |||||||
| Current Taxes | $ | (48,267 | ) | $ | (60,461 | ) | ||
| Deferred Taxes | - | - | ||||||
| Income Taxes Expense (Benefit) | $ | (48,267 | ) | $ | (60,461 | ) | ||
The items accounting for the difference between income taxes at the Australia statutory rate and the provision for income taxes are as follows:
| Year Ended | ||||||||||||||||
| June 30, | June 30, | |||||||||||||||
| 2014 | 2013 | |||||||||||||||
| Amount | Impact on Rate | Amount | Impact on Rate | |||||||||||||
| Income Tax Expense (Benefit) at Australia Statutory Rate | $ | (274,229 | ) | (31.24 | )% | $ | (492,334 | ) | (32.75 | )% | ||||||
| Expenses paid by parent on behalf of foreign subsidiary | 92,480 | 10.54 | % | 351,935 | 23.41 | % | ||||||||||
| R&D Refundable Tax Credit | (48,267 | ) | (5.50 | )% | (60,461 | ) | (4.02 | )% | ||||||||
| Reduction of NOL Carryforward Due to R&D Tax Credit | 48,267 | 5.50 | % | 60,461 | 4.02 | % | ||||||||||
| Change in deferred Tax Valuation Allowance | 260,533 | 29.68 | % | (363,703 | ) | (24.20 | )% | |||||||||
| Foreign Exchange Rate Changes | (127,051 | ) | (14.47 | )% | 443,641 | 29.52 | % | |||||||||
| Total Income Tax Expense (Benefit) | $ | (48,267 | ) | (5.50 | )% | $ | (60,461 | ) | (4.02 | )% | ||||||
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and amounts used for income tax purposes. Significant components of the Company's net deferred income taxes are as follows:
| F-16 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
| June 30, | June 30, | |||||||
| 2014 | 2013 | |||||||
| Current Deferred Tax Assets | ||||||||
| Warrant derivative liability | $ | 28,209 | $ | - | ||||
| Provision for annual leave | 18,848 | 14,813 | ||||||
| Superannuation | 3,815 | 3,699 | ||||||
| Total Current Deferred Tax Assets | $ | 50,872 | $ | 18,512 | ||||
| Current Deferred Tax Liabilities | ||||||||
| Prepaid Investor Services | $ | - | $ | - | ||||
| Prepaid expenses | - | - | ||||||
| Prepaid insurance | - | - | ||||||
| Accounts Payable/trade creditors | - | - | ||||||
| Patent Costs | - | - | ||||||
| Total Current Deferred Tax Liabilities | $ | - | $ | - | ||||
| Non-Current Deferred Tax Assets | ||||||||
| Prepaid Investor Services | $ | 751,917 | $ | 1,393,309 | ||||
| Capital Raising Costs | 28,622 | 27,752 | ||||||
| Legal Costs | 29,019 | 28,337 | ||||||
| Intellectual Property | 14,288 | 13,854 | ||||||
| Patent Costs | 59,473 | 32,425 | ||||||
| Formation Expense | 8,757 | 8,491 | ||||||
| Net Operating Loss Carryover | 3,259,060 | 2,418,795 | ||||||
| Foreign Exchange Loss (OCI) | 90,859 | 73,377 | ||||||
| Total Non-Current Deferred Tax Assets | 4,241,995 | 3,996,340 | ||||||
| Deferred Tax Valuation Allowance | (4,292,867 | ) | (4,014,852 | ) | ||||
| Total Non-Current Deferred Tax Assets | (50,872 | ) | (18,512 | ) | ||||
| Total Deferred Tax Assets (Net) | $ | - | $ | - | ||||
Management has determined that the realization of the net deferred tax asset is not assured and has created a valuation allowance for the entire amount of such benefits.
The Company follows ASC 740-10, which provides guidance for the recognition and measurement of certain tax positions in an enterprise’s financial statements. Recognition involves a determination whether it is more likely than not that a tax position will be sustained upon examination with the presumption that the tax position will be examined by the appropriate taxing authority having full knowledge of all relevant information.
The Company’s policy is to record interest and penalties associated with unrecognized tax benefits as additional income taxes in the statement of operations. As of June 30, 2014 the Company had no unrecognized tax benefits. There were no changes in the Company’s unrecognized tax benefits during the years ended June 30, 2014 and 2013. The Company did not recognize any interest or penalties during fiscal 2014 or 2013 related to unrecognized tax benefits.
The income tax returns filed for the tax years from inception will be subject to examination by the relevant taxing authorities.
NOTE 8 – STOCKHOLDERS’ DEFICIT
In September 2012, the board authorized additional share issuances to three investors who previously converted convertible debentures at $1.50 per share per the terms of the debentures. The additional share issuance was to ratchet the prior conversions from $1.50 per share, down to $0.65 per share. As a result, the Company issued 147,052 additional shares of common stock valued at $0.65, based on contemporaneous cash offering prices, and recorded an expense of $95,611 as the original agreement didn't call for price protection.
In September 2012, a $75,000 convertible debenture was converted into shares of common stock pursuant to a conversion notice. $76,896 of principal and interest was converted at $1.50 into 51,264 shares. The original agreement stipulated a conversion price of $1.50 however, as the Company voluntary ratcheted down the conversion to $0.65, the Company recorded an additional expense of $43,547 (based on contemporaneous cash sales prices of $0.65) related to the additional 67,037 shares issued.
In September 2012, the Company entered into an agreement to issue 300,000 shares of common stock for services rendered during the three months ended September 30, 2012. The shares were valued at $0.65 based on contemporaneous cash offering prices and accordingly, the Company recognized an expense of $195,000.
| F-17 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
In September 2012, the Company issued 30,000 shares of common stock for past services to a consultant. The shares were valued at $0.65 based on contemporaneous cash offering prices and accordingly, the Company recognized an expense of $19,500 related to the transaction.
In October 2012, the Company entered into an agreement to issue 300,000 shares of common stock for services rendered during the three months ended December 31, 2012. The shares were valued at $0.65 based on contemporaneous cash offering prices and accordingly, the Company recognized an expense of $195,000.
In November 2012, the Company entered into an agreement to issue 35,000 shares of common stock for services rendered during the three months ended December 31, 2012. The shares were valued at $0.65 based on contemporaneous cash offering prices and accordingly, the Company recognized an expense of $22,750.
In November 2012, the Company entered into an agreement to issue 50,000 shares of common stock for services rendered during the three months ended December 31, 2012. The shares were valued at $0.65 based on contemporaneous cash offering prices and accordingly, the Company recognized an expense of $32,500.
In November 2012, the Company issued 100,000 shares to settle approximately $21,000 of accounts payable. The Company recorded a loss on settlement of approximately $43,000 as the shares were valued at $0.65 per share or $65,000 based on contemporaneous cash offering prices.
In December 2012, the Company issued 10,000 shares of common stock for past services to a consultant. The shares were valued at $0.65 based on contemporaneous cash offering prices and accordingly, the Company recognized an expense of $6,500 related to the transaction.
In December 2012, the Company entered into an agreement to issue 50,000 shares of common stock for services rendered during the three months ended December 31, 2012. The shares were valued at $0.65 based on contemporaneous cash offering prices and accordingly, the Company recognized an expense of $32,500.
In December 2012, a total of 1,021,460 shares were returned to the Company in a settlement with a shareholder.
In March 2013, the Company entered into an agreement to issue 25,000 shares of common stock for services rendered during the three months ended March 31, 2013. The shares were valued at $0.65 based on contemporaneous cash offering prices and accordingly, the Company recognized an expense of $16,250.
In March 2013, the Company entered into an agreement to issue 125,000 shares of common stock to settle approximately $16,500 of accounts payable. The Company recorded a loss on settlement of approximately $65,000 as the shares were valued at $0.65 per share or $81,250 based on contemporaneous cash offering prices.
In March 2013, the Company entered into an agreement to issue 7,500 shares of common stock for services rendered during the three months ended March 31, 2013. The shares were valued at $0.65 per share or $4,875 based on contemporaneous cash offering prices and accordingly, the Company recognized an expense of $4,875.
In March 2013, a total of 2,560,571 shares were returned to the Company in a settlement with a shareholder.
In March 2013, the Company entered into an agreement to issue 200,000 shares of common stock for services rendered during the three months ended March 31, 2013. The shares were valued at $0.65 per share or $130,000 based on contemporaneous cash offering prices and accordingly, the Company recognized an expense of $130,000.
In March 2013, the Company entered into an agreement to issue 5,000 shares of common stock for services rendered during the three months ended March 31, 2013. The shares were valued at $0.65 per share or $3,250 based on contemporaneous cash offering prices and accordingly, the Company recognized an expense of $3,250.
| F-18 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
In May 2013, the Company became obligated to issue 25,000 shares of common stock for services rendered by a consultant. These shares were valued at $0.20 per share or $5,000 based on the quoted market price of the stock on the date of the grant. These shares are reflected as common stock issuable and the Company recognized an expense of $5,000.
On June 6, 2013, the Company entered into a 60 day agreement with a consultant whereby they would issue that consultant 600,000 shares of the Company's common stock. These shares were valued at $0.20 per share or $120,000 based on the quoted market price of the stock on the date of the grant. The Company is recognizing the expense pro rata over the 60 day term. As of June 30, 2013 the Company recognized $48,000 with a credit to additional paid-in-capital.
In July 2013, the Company issued 300,000 shares of common stock to a consultant related to a June 6, 2013 agreement. The shares were valued at $0.20 per share (based on current market price) and accordingly, the Company recognized an expense of $12,000 during the first quarter of fiscal 2014 and $48,000 was previously recognized during fiscal 2013 as the expense was amortized over the term of the agreement.
In July 2013, the Company issued 250,000 shares of common stock to a consultant for past services. The shares are fully vested and valued at $0.20 per share (based on current market price) and accordingly, the Company recognized an expense of $50,000 related to the share issuance.
In July 2013, the Company issued 137,500 shares of common stock to a consultant in exchange for a $27,500 accounts payable relating to past services. The shares are fully vested and valued at $0.20 per share (based on current market price) and accordingly there was no gain or loss on this settlement.
In July 2013, the Company issued 10,000 shares of common stock to a consultant for past services. The shares are fully vested and valued at $0.20 per share (based on current market price) and accordingly, the Company recognized an expense of $2,000 related to the share issuance.
In July 2013, the Company issued 150,000 shares of common stock to a consultant for past services. The shares are fully vested and valued at $0.20 per share (based on current market price) and accordingly, the Company recognized an expense of $30,000 related to the share issuance.
In September 2013, the Company issued the balance of 300,000 shares of common stock to a consultant related to a June 6, 2013 agreement. The shares were valued at $0.20 per share (based on current market price) and accordingly, the Company recognized an expense of $60,000 during the three months ended September 30, 2013.
On September 30, 2013, pursuant to a consulting agreement, the company issued 25,000 shares of common stock for past services performed during the quarter. The shares were valued at $0.20 per share (based on current market price) and accordingly, the Company recognized an expense of $5,000 during the three months ended September 30, 2013.
In October 2013, the Company issued 500,000 vested shares of common stock as a non-refundable retainer in conjunction with a 90-day investment banking services agreement. The shares were valued at the market price on the day of the grant, $0.10, and the Company recorded an expense of $50,000.
In October 2013, the Company issued 200,000 shares of common stock to a consultant for services. The shares were issued to the consultant and vest over the three year term of the agreement. The shares were valued at $0.10 per share (based on current market price) and accordingly, the Company recognized an expense of $13,333 and has recorded $6,667 in prepaid expenses for January's services.
In October 2013, the Company issued 100,000 shares of common stock to a consultant for past services. The shares are fully vested and valued at $0.10 per share (based on current market price) and accordingly, the Company recognized an expense of $10,000 related to the share issuance.
| F-19 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
In November 2013, the Company issued 30,000 shares of common stock to a consultant for past services. The shares are fully vested and valued at $0.10 per share (based on current market price) and accordingly, the Company recognized an expense of $3,000 related to the share issuance.
In November 2013, the Company issued 25,000 shares of common stock to a consultant for past services. The shares are fully vested and valued at $0.10 per share (based on current market price) and accordingly, the Company recognized an expense of $2,500 related to the share issuance.
On May 9, 2014, the Company entered into an agreement with a consultant to provide services over a six month period. The Company agreed to issue the consultant 500,000 shares of common stock upon signing the agreement and 500,000 shares at the end of the six months. No shares were issued as of June 30, 2014 and accordingly the Company valued the 1,000,000 shares based on the market price on the agreement date of $0.10 and will recognize the resulting $100,000 of expense through November 2014.
Warrants:
In September, 2013, pursuant to convertible debenture, the Company issued 3,000,000 warrants to purchase common stock. These warrants have an initial exercise price of $0.0698 per share which is subject to adjustment and expire 5 years from the date of issuance (See Note 6).
NOTE 9 – COMMITMENTS AND CONTINGIENCIES
Legal Matters
From time to time, we may be involved in litigation relating to claims arising out of our operations in the normal course of business. As of June 30, 2014, there were no pending or threatened lawsuits that could reasonably be expected to have a material effect on the results of our operations.
Operating Agreements
In November 2009, the Company entered into a commercialization agreement whereby the Company agreed to pay royalties of 2% of net revenues. Additionally, the Company agreed to pay 5% of each and every license agreement subscribed for. The contract is cancellable at any time by either party. To date, no amounts are owed under the agreement.
Operating Leases
In September 2009, the Company entered into a month to month lease agreement with monthly rent at $1,016 per month which in fiscal 2012, became subject to a 3.5% escalation clause or $1,052 per month. In July 2013, the Company moved to new premises. No formal agreement has been entered regarding leasing of the new office space and no amounts have been paid, but an accrued liability at June 30, 2014 of $11,303 has been recognized in anticipation of a month to month agreement retroactive to July 1, 2013 at $942 per month.
Rent expense for the years ended June 30, 2014 and 2013 were $11,016 and $13,074 respectively.
NOTE 10 – RELATED PARTY TRANSACTIONS
Since inception, Propanc Health Group Corporation has conducted transactions with directors and director related entities. These transactions included the following:
| F-20 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
As of June 30, 2014 and 2013, the Company owed certain directors a total of $161,975 and $130,689 respectively, for money loaned to the Company throughout the years. The loan balance owed at June 30, 2014 was not interest bearing.
As of June 30, 2014 and 2013, the Company owed two directors a total of $60,350 and $57,237, respectively, related to expenses incurred on behalf of the Company related to corporate startup costs and intellectual property.
NOTE 11 – CONCENTRATIONS AND RISKS
Concentration of Credit Risk
The Company maintains its cash in bank and financial institution deposits in Australia. Bank deposits in Australian banks are uninsured. The Company has not experienced any losses in such accounts through June 30, 2014.
Receivable Concentration
As of June 30, 2014 and 2013, the company's receivables were 100% related to reimbursements on GST taxes paid.
Vendor Concentration
As of June 30, 2014, there was one significant vendors that the Company relies upon to conduct its research and development. This vendor provided services to the Company which can be replaced by alternative vendors should the need arise.
Product and Patent Concentration
As of June 30, 2014 the Company was undertaking preclinical activities for their lead product. The Company was also undertaking research to uncover the mechanism of action of their lead product in order to screen new compounds for development.
The Company previously expanded by the filing of an international PCT patent application (No. PCT/AU2010/001403) directed to enhanced proenzyme formulations and combination therapies. The international PCT application has been based on previous provisional patent applications capturing the Company’s ongoing research and development in this area.
The Company received grant status in South Africa and more recently in New Zealand. In addition, the United States Patent and Trademark Office or USPTO and European Patent Office or EPO have made preliminary indications that key features of our technology are patentable. The Company is presently working towards securing a patent in each region, covering as many aspects of its technology as possible, whilst also actively seeking protection throughout Eastern Europe, Asia and South America.
Individual countries and regions, include United States, Canada, Japan, Brazil, China, Mexico, Hong Kong, Singapore, Israel, Chile, Peru, Malaysia, Vietnam, Indonesia, Europe, Russia, India, Australia and South Korea. The patent is granted in South Africa and New Zealand.
Further provisional patent filings are also expected to be filed to capture and protect additional patentable subject matter that is identified, namely further enhanced formulations, combination treatments, use of recombinant products, modes of action and molecular targets.
Foreign Operations
As of June 30, 2014 and 2013, the Company's operations are based in Australia.
| F-21 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
NOTE 12 - DERIVATIVE FINANCIAL INSTRUMENTS and FAIR VALUE MEASUREMENTS
Derivative Financial Instruments:
The Company applies the provisions of ASC Topic 815-40, Contracts in Entity’s Own Equity (“ASC Topic 815-40”), under which convertible instruments and warrants, which contain terms that protect holders from declines in the stock price (reset provisions), may not be exempt from derivative accounting treatment. As a result, warrants are recorded as a liability and are revalued at fair value at each reporting date. If the fair value of the warrants exceeds the face value of the related debt, the excess is recorded as change in fair value in operations on the issuance date. The Company has 3,000,000 warrants with repricing options outstanding at June 30, 2014.
The Company calculates the estimated fair values of the liabilities for warrant derivative instruments using the Black Scholes (BSM) option pricing model. The closing price of the Company’s common stock at September 30, 2013 was $0.20, while the closing price of the Company’s common stock at June 30, 2014 was $0.09. Volatility, expected term and risk free interest rates used to estimate the fair value of derivative liabilities at June 30, 2014, are indicated in the table that follows. The volatility was based on comparative company’s methods since the Company’s stock is very thinly traded, the expected term is equal to the remaining term of the warrants and the risk free rate is based upon rates for treasury securities with the same term.
Warrants
| Initial Valuation September 30, 2013 | June 30, 2014 | |||||||
| Volatility | 53 | % | 134 | % | ||||
| Expected Term | 2.25 | 1.5 | ||||||
| Risk Free Interest Rate | 0.4 | % | 0.47 | % | ||||
| Expected dividend yield | none | none | ||||||
Fair Value Measurements:
We currently measure and report at fair value the liability for warrant derivative instruments. The fair value liabilities for price adjustable warrants have been recorded as determined utilizing the BSM option pricing model. The following tables summarize our financial assets and liabilities measured at fair value on a recurring basis as of June 30, 2014:
| Quoted Prices | Significant | |||||||||||||||
| Balance at | in Active | Other | Significant | |||||||||||||
| June 30, | Markets for | Observable | Unobservable | |||||||||||||
| 2014 | Identical Assets | Inputs | Inputs | |||||||||||||
| (Level 1) | (Level 2) | (Level 3) | ||||||||||||||
| Fair value of liability for warrant derivative instruments | $ | 158,244 | $ | — | $ | — | $ | 158,244 | ||||||||
| F-22 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
The following is a roll forward for the year ended June 30, 2014 of the fair value liability of price adjustable warrant derivative instruments:
| Fair Value of | ||||
| Liability for | ||||
| Warrant | ||||
| Derivative | ||||
| Instruments | ||||
| Balance at June 30, 2013 | $ | - | ||
| Initial fair value recording of warrant derivative liability as debt discount | 144,241 | |||
| Effects of foreign currency exchange rate changes | (2,519 | ) | ||
| Change in fair value included in statements of operations | 16,522 | |||
| Balance at June 30, 2014 | $ | 158,244 | ||
NOTE 13 – SUBSEQUENT EVENTS
Conversion of Debentures
On July 2, 2014, the $139,680 convertible debenture and accrued interest was converted using the contractual conversion rate into 2,183,333 shares of the Company’s common stock.
Settlement and Stipulation Agreement
In July 2014, the Company signed a term sheet and a Settlement and Stipulation Agreement (the "Settlement Agreement") with a third party purchaser (the "purchaser") to have that purchaser acquire certain portions of the Company’s liabilities to creditors (“Creditors”) in exchange for an obligation of the Company to issue shares of common stock to the purchaser, which shares of common stock would then be sold by the purchaser and 65% of the net proceeds, as defined, distributed to the Creditors. The shares are to be freely traded shares issued pursuant to section 3(a)(10) of the Securities Act of 1933.
Under the terms of the Settlement Agreement, the variable quantity of common stock would be issued in tranches such that the purchaser would not own more than 9.99% of the outstanding shares of common stock at any time.
Under the above agreements, in May 2014 the Company also paid an expense fee of $25,000 in the form of a convertible promissory note. (see Note 6)
The purchaser entered into agreements through July 2014 with the Creditors to acquire $627,998 in liabilities of the Company and filed a complaint with the Second Judicial Circuit Court in Leon County, Florida seeking a judgment against the Company for such amount. A court order based on this complaint was issued on September 9, 2014, (the "court order date") resulting in the transfer of $627,998 in liabilities of the Company to the purchaser. In addition, upon entry of the order, the Company became obligated to issue the purchaser a purchaser fee of $50,000 worth of common stock priced at 75% of the average closing bid prices for the 10 days immediately preceding the date of the order. As a result of the purchased liabilities and purchaser fee, the Company became obligated to issue to the purchaser approximately $1,034,000 worth of common stock. These liabilities now meet the criteria of stock settled debt under ASC 480 resulting in the recording of a liability premium of approximately $356,000 with a charge to interest expense on the court order date.
The Company issued an initial tranche of 7,426,000 shares of common stock to the purchaser in September 2014.
| F-23 |
PROPANC HEALTH GROUP CORPORATION AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2014 and 2013
Equity Purchase Agreement
On July 18, 2014 the Company executed an Equity Purchase Agreement (the "agreement") with an investor (the "investor") affiliated with the above purchaser. The Company may sell (put shares) from time to time, during the commitment period discussed below, up to $5,000,000 of the Company's common stock at a sale price equal to 90% of the of the market price. The market price is determined during a valuation period which is the 10 trading days immediately following the clearing date (the date when the put shares are deposited into the investor's brokerage account) associated with the applicable put notice. The valuation period may change based on any valuation events occurring, as defined in the agreement. The Company's right to sell to the investor and the investor's obligation to purchase shares is subject to certain restrictions, including a floor price, as defined in the agreement. Furthermore, on each closing date the number of shares then to be purchased shall not exceed that amount that when aggregated with all other shares beneficially owned by the investor would result in the investor owning more than 9.99% of the outstanding shares of common stock.
The commitment period is the earlier of the sale of $5,000,000 worth of shares or 24 months.
On July 18, 2014, Company entered into the Registration Rights Agreement with the investor. Pursuant to the terms of the Registration Rights Agreement, the Company is obligated to file a registration statement (the “Registration Statement”) with the SEC to cover the Registrable Securities within one hundred twenty (120) days of closing. The Company must use its commercially reasonable efforts to cause the Registration Statement relating to the Registered Securities to become effective within five (5) business days after notice from the SEC that such Registration Statement may be declared effective, and keep the Registration Statement effective at all time prior to the termination of the Equity Purchase Agreement until the earliest of (i) date that is three months after the completion of the last Closing date (as defined in the Equity Purchase Agreement), (ii) the date when the investor may sell all Registered Securities under Rule 144 without volume limitations, or (iii) the date the investor no longer owns any of the Registered Securities (collectively, the “Registration Period”).
On July 18, 2014 the Company paid a $50,000 fee to the investor in the form of a $50,000 promissory note, non-bearing interest and due January 31, 2015.
Convertible Promissory Note
On August 6, 2014 (execution date), the Company executed a convertible promissory note in the principal sum of $250,000, with an original issue discount of $25,000. The consideration to be paid to the Lender shall be equal to the consideration actually paid by the Lender plus prorated interest and any other fees such that the Company shall be required to pay. The original issue discount shall also be prorated based on the actual consideration received to equal approximately 10% of the consideration received. If the Company repays a consideration payment on or before the first 90 days from the effective date of that payment, the interest rate on that payment of consideration will be 0%. If the company does not repay a payment on or before the 90 days, the Company will incur a one-time interest charge of 12% on the principal amount of each loan. Upon execution of the note, the note holder made an initial payment of $25,000 to the Company of the total consideration. The maturity date is two years from the date of each payment to the Company, and is the date upon which the principal sum, as well as any unpaid interest and other fees, shall be due and payable. The note is convertible, at the option of the investor, to common stock of the Company at any time after the effective date at the lesser of $0.09 or 60% of the lowest trade price in the 25 trading days previous to the conversion. This note will be bifurcated with the embedded conversion option recorded as a derivative liability at fair value.
| F-24 |
Item 9. Changes In and Disagreements with Accountants on Accounting and Financial Disclosure.
None
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Our management is responsible for establishing and maintaining disclosure controls and procedures that are designed to ensure that information required to be disclosed in our reports under the Securities Exchange Act of 1934 (the “Exchange Act”) is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure based closely on the definition of “disclosure controls and procedures” in Rule 15d-15(e) under the Exchange Act. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
At the end of the period covered by this Annual Report, we conducted an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures. Based upon the foregoing, our Chief Executive Officer and Chief Financial Officer concluded that, as of June 30, 2014, the disclosure controls and procedures of our Company were not effective to ensure that the information required to be disclosed in our Exchange Act reports was recorded, processed, summarized and reported on a timely basis.
The Company is undertaking to improve its internal control over financial reporting and improve its disclosure controls and procedures. As of June 30, 2014, we had identified the following material weaknesses which still exist through the date of this report:
| 38 |
As of June 30, 2014 and as of the date of this report, we did not maintain effective controls over the control environment. Specifically, the Board does not currently have a director who qualifies as an audit committee financial expert as defined in Item 407(d)(5)(ii) of Regulation S-K. The Company also lacks accounting personnel with technical knowledge in certain debt and equity transactions. Additionally, because of the size of the Company’s administrative staff, controls related to the segregation of certain duties have not been developed and the Company has not been able to adhere to them. Since these entity level programs have a pervasive effect across the organization, management has determined that these circumstances constitute a material weakness.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). The design of any system of controls is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions, regardless of how remote. All internal control systems, no matter how well designed, have inherent limitations. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
We carried out an evaluation, under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, of the effectiveness of our internal controls over financial reporting as of June 30, 2014. Based on this assessment, management believes that, as of June 30, 2014, we did not maintain effective controls over the financial reporting control environment. Specifically, the Board does not currently have a director who qualifies as an audit committee financial expert as defined in Item 407(d)(5)(ii) of Regulation S-K. Further, because of the limited size of its administrative support staff, and due to the financial constraints on the Company, management has not been able to develop or implement controls related to the segregation of duties for purposes of financial reporting. The Company also lacks accounting personnel with technical knowledge in certain debt and equity transactions. Because of these material weaknesses, management has concluded that we did not maintain effective internal control over financial reporting as of June 30, 2014, based on the criteria established in the “Internal Integrated Framework” issued by COSO.
No Attestation Report by Independent Registered Accountant
The effectiveness of our internal control over financial reporting as of June 30, 2014 has not been audited by our independent registered public accounting firm by virtue of our exemption from such requirement as a smaller reporting company.
Changes in Internal Controls over Financial Reporting
There were no changes in internal controls over financial reporting that occurred during the period covered by this report, which have materially affected, or are reasonably likely to materially affect, our internal controls over financial reporting.
Corrective Action
Our Board is seeking a candidate with audit committee financial expertise to serve as an independent director of the Company and as the Chairman of our audit committee. Management also plans to make future investments in the continuing education of our accounting and financial staff. Improvements in our disclosure controls and procedures and in our internal control over financial reporting will, however, depend on our ability to add additional resources and independent directors to provide more internal checks and balances, and to provide qualified independence for our audit committee. We believe we will be able to commence achieving these goals once our sales and cash flow grow and our financial condition improves.
| 39 |
None.
Item 10. Directors, Executive Officers, and Corporate Governance
The following is a list of our directors and executive officers. All directors serve one-year terms or until each of their successors are duly qualified and elected. The officers are elected by our Board.
| Name | Age | Position | ||||
| James Nathanielsz | 40 | Chief Executive Officer, Secretary, Treasurer and Director | ||||
| Dr. Julian Kenyon | 67 | Director | ||||
James Nathanielsz has served as a director since inception. Mr. Nathanielsz has served as a director and Chief Executive Officer of our Australian company since October 2007. From July 2006 until October 2007, Mr. Nathanielsz served as the New Products Manager of Biota Holdings Limited, an anti-infective drug development company in Australia. Mr. Nathanielsz was selected as a director because he is the Co-Founder of our Australian company and for his experience in R&D and manufacturing and distribution. Mr. Nathanielsz graduated with a Bachelor of Applied Science, majoring in Biochemistry/Applied Chemistry and subsequently with a Master of Entrepreneurship & Innovation from Swinburne University of Technology in Melbourne, Australia.
Dr. Julian Kenyon has served as a director since inception. Dr. Kenyon founded our Australian company and was appointed as a director of our Australian company on February 12, 2008. Since 2000, Dr. Kenyon has served as an integrated medical physician and Medical Director of the Dove Clinic for Integrated Medicine in Winchester and London. Dr. Kenyon is the Founder-Chairman of the British Medical Acupuncture Society in 1980 and Co-Founder of the Centre for the Study of Complementary Medicine in Southampton and London. Dr. Kenyon was selected as a director because he is the Co-Founder of the Australian subsidiary and the business is based on his initial work at the Dove Clinic. Dr. Kenyon graduated from the University of Liverpool with a Bachelor of Medicine and Surgery and subsequently with a research degree, Doctor of Medicine. Since 1972, he was appointed a Primary Fellow of the Royal College of Surgeons, Edinburgh.
Committees of the Board of Directors
We presently do not have an audit committee, nominating committee, compensation committee, or other committee or committees performing similar functions, as our management believe that until this point it has been premature at the early stage of our management and business development to form an audit, compensation or other committees.
Scientific Advisory Board
We have a Scientific Advisory Board that provides advice relating to the following:
| · | The identification, assessment, evaluation, selection, conduct and management of research projects, both those which are under review and are in progress; |
| · | Intellectual property; and |
| · | Commercialization. |
| 40 |
The Scientific Advisory Board may also address issues related to improving project selection, formal review processes and management procedures within Propanc Health Group. The Scientific Advisory Board will generally be composed of an advisory panel of clinicians with expertise in translational research.
As of September 30, 2014, the members of the Scientific Advisory Board were:
| · | Professor John Smyth |
| · | Professor Klaus Kutz (Acting Chief Medical Officer, Propanc Health Group) |
| · | Professor Karrar Khan |
| · | Dr. Ralf Brandt |
Each of the members of our Scientific Advisory Board acts as an independent consultant and is compensated on an hourly basis for his services. There is presently no stock based compensation for their services.
Professor Kutz is also acting as Chief Medical Officer for Propanc, His compensation continues to be based on an hourly rate as per his Advisory Board Agreement. Propanc intends to appoint Professor Kutz as Chief Medical Officer for Propanc in a full time capacity at a time which is mutually agreed upon between both parties.
Professor John Smyth
John Smyth has for the past 25 years served as Chair of Medical Oncology in the University of Edinburgh Medical School, where his major research interest is the development and evaluation of new anti-cancer drugs. He has published over 300 papers and is Editor-in-Chief of the European Journal of Cancer. He served for several years on the UK Committee on Safety of Medicines; currently Chair's the Expert Advisory Group for Oncology & Haematology for the Commission on Human Medicines and serves on the Expert Oncology Advisory Group to the European Drug Licensing Board. He is a fellow of the Royal College of Physicians of Edinburgh and London, and fellow of the Royal Society of Edinburgh. He is a past-president of the European Society of Medical Oncology and was from 2005 - 2007 President of the Federation of European Cancer Societies.
Professor Klaus Kutz
Professor Kutz has fifteen years of experience as independent consultant in Clinical Pharmacology and Safety for pharmaceutical companies and clinical research organizations. His specialty over the last six years is Oncology, including preparation of multiple NDAs and INDs for small and medium sized pharmaceutical companies. He has prepared, organized and reported clinical Phase I studies in oncology and Phase II studies in different cancer indications (prostate, gastric, ovarian, small cell lung cancer) and Non-Hodgkin Lymphomas. Professor Kutz has more than 12 years of experience as Head of Clinical Pharmacology with world-wide responsibilities for Phase I and Clinical Pharmacokinetics in two internationally operating pharmaceutical companies, setting up and restructuring international Clinical Pharmacology departments. His achievements include the successful world-wide registration of multiple important Sandoz’ compounds by preparation of multiple NDAs (New Drug Applications) and Expert reports (including Written Summary), as well as the preparation of multiple INDs (Investigational New Drug Applications) for Sandoz Pharma Ltd and Sanofi Research. A specialist for Internal Medicine, Gastroenterology, and Clinical Pharmacology, he is also Professor of Medicine at the University of Bonn, Germany.
| 41 |
Professor Karrar Khan
Professor Khan has over 35 years of experience in drug discovery, pharmaceutical development, registration and management of pharmaceutical scientists. Professor Khan has also held various product development and management positions with Abbott Laboratories and Beecham Pharmaceuticals. In these roles, he developed medicines for several therapeutic areas including antibiotics, anti-depressant, anti-inflammatory, anti-obesity, psychosis, cardiovascular, pain, cancer, Parkinson’s disease and diabetes. Professor Khan developed and contributed to the launch of two once a day controlled release dosage forms. His expertise ranged from development for phase 1 to phase 3- 4 and significant experience of bringing prescription and OTC products to market on a worldwide bases (contributed to the registration and launch of over 60 pharmaceutical products). He is a qualified person under the EC quality assurance directive. He now works as a pharmaceutical development consultant. Professor Khan has authored or co-authored more than 40 scientific publications and is an inventor of several development patents. He has been an invited speaker at many national and international conferences.
Dr. Ralf Brandt
Dr. Brandt is the co-founder of vivoPharm. He is a biochemist and cell biologist with over 15 years of experience in research programs of experimental oncology. Furthermore, he has immense experience in in vivo pharmacology and anti-cancer drug profiling. He received his License (BSc in Biochemistry and Animal Physiology) in 1986, and his PhD (in Biochemistry) in 1991 from the Martin-Luther University of Halle-Wittenberg, Germany. Dr. Brandt was employed at research positions at the National Cancer Institute in Bethesda, MD, USA and at Schering AG, Germany. Since 1990, Dr. Brandt has been active in the field of preclinical oncology. He led the Tumour Biology program at Novartis Pharma AG, Switzerland and established several transgenic mouse lines developing tumors under the control of oncogenes. During Dr. Brandt's long career in the pharmaceutical industry he has acquired significant knowledge and expertise in leading business units and representation of services to the pre-clinical research market. Dr. Brandt is a member of the Scientific Advisory Board at Receptor Inc. in Toronto Canada.
Code of Ethics
The Board is currently reviewing a Code of Conduct and Ethics (the “Code”) to apply to all of our directors, officers and employees. The Code is intended to promote ethical conduct and compliance with laws and regulations, to provide guidance with respect to the handling of ethical issues, to implement mechanisms to report unethical conduct, to foster a culture of honesty and accountability, to deter wrongdoing and to ensure fair and accurate financial reporting. Upon approval by the Board, a copy of the Code will be available at our website www.propanc.com
Shareholder Communications
Although we do not have a formal policy regarding communications with the Board, shareholders may communicate with the Board by writing to us at Level 13, Suite 1307, 530 Collins Street, Melbourne, VIC, 3000, Australia, Attention: Corporate Secretary, or by facsimile +61 (0) 3 9614 7194. Shareholders who would like their submission directed to a member of the Board may so specify, and the communication will be forwarded, as appropriate.
Board Diversity
While we do not have a formal policy on diversity, our Board considers diversity to include the skill set, background, reputation, type and length of business experience of our Board members as well as a particular nominee’s contributions to that mix. Our Board believes that diversity brings a variety of ideas, judgments and considerations that benefit Propanc and our shareholders. Although there are many other factors, the Board seeks individuals with experience in business, financial and scientific research and development.
Board Assessment of Risk
Our risk management function is overseen by our Board. Our management keeps our Board apprised of material risks and provides our directors access to all information necessary for them to understand and evaluate how these risks interrelate, how they affect Propanc, and how management addresses those risks. Mr. Nathanielsz, as our Chief Executive Officer works closely together with the Board once material risks are identified on how to best address such risk. If the identified risk poses an actual or potential conflict with management, our independent directors may conduct the assessment. Presently, the primary risks affecting Propanc is the lack of working capital, the inability to generate sufficient revenues so that we have positive cash flow from operations and success of future clinical trials. The Board focuses on these key risks at each meeting and actively interfaces with management on seeking solutions.
| 42 |
Item 11. Executive Compensation.
Termination Provisions
Upon termination by Propanc and in accordance with Mr. Nathanielsz employment agreement, Mr. Nathanielsz is entitled to six months of base salary. Upon his resignation, Mr. Nathanielsz is entitled to twelve (12) weeks of base salary.
Summary Compensation Table
The following table sets forth the compensation paid or accrued by us to our Chief Executive Officer, Chief Financial Officer and each of our other officers for the years ended June 30, 2014 and 2013.
Summary Compensation Table for Fiscal 2014 and 2013
| Name and Principal Position | Year | Salary ($) | All Other Compensation ($) | Total ($) | ||||||||||||
| James Nathanielsz (1) | 2014 | 137,685 | 12,736 | (2) | 150,421 | |||||||||||
| Chief Executive Officer | 2013 | 154,035 | 13,863 | (2) | 167,898 | |||||||||||
| Dr. Klaus Kutz (3) | 2014 | 7,936 | - | 7,936 | ||||||||||||
| Acting Chief Medical Officer | 2013 | 4,261 | - | 4,261 | ||||||||||||
_________
(1) Under an employment agreement dated August 15, 2010, Mr. Nathanielsz receives a gross annual salary of $150,000 AUD per year.
(2) Under the employment agreement with the Company, Mr. Nathanielsz receives a a 9.25% contribution to a pension of which he is the beneficiary.
(3) Pursuant to certain mutual agreement with the Company, Dr. Kutz is compensated on per diem basis for his services rendered until the Company secures sufficient fund to employ Dr. Kutz on full-time basis.
Outstanding Equity Awards
There are no outstanding equity awards.
Equity Compensation Plan Information
We currently do not have an equity compensation plan.
Director Compensation
We do not pay cash compensation to our directors for service on our Board and our employees do not receive compensation for serving as members of our Board. Directors are reimbursed for reasonable expenses incurred in attending meetings and carrying out duties as board members.
Section 16(a) Beneficial Ownership Reporting Compliance
Under the securities laws of the United States, our directors, executive (and certain other) officers, and any persons holding ten percent or more of our Common Stock must report on their ownership of the Common Stock and any changes in that ownership to the Commission. Specific due dates for these reports have been established. During the fiscal year ended June 30, 2014, we believe that all reports required to be filed by Section 16(a) were filed on a timely basis.
| 43 |
Insider Trading Policy
The Company's Board is currently reviewing an insider trading policy (the “Insider Trading Policy”) that will establish guidelines and procedures for the trading of Company securities by officers, directors, employees and consultants (“Insiders”). Among others, the Insider Trading Policy shall establish prohibitions on insider trading, tipping, short term trading and short sales; provides for quarterly black-out restrictions on trading and guidelines for establishment of Rule 10b5-1 trading plans. The Insider Trading Policy shall encourage Insiders who wish to trade in Company securities to consult with the General Counsel of the Company prior to trading. Upon approval by the Board, the Insider Trading Policy will be available on our website at www.propanc.com.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The following table sets forth the number of shares of our voting stock beneficially owned, as of October 14, 2014 by (i) those persons known by Propanc to be owners of more than 5% of Propanc’s common stock, (ii) each director, (iii) our Named Executive Officer, and (iv) all executive officers and directors as a group:
| Title of Class | Name and Address of Beneficial Owner | Amount and Nature of Beneficial Owner(1) | Percent of Class (1) | |||||||
| Common Stock | James Nathanielsz 576 Swan Street Richmond, VIC, 3121, Australia (2) | 9,701,761 | 11.77 | % | ||||||
| Common Stock | Dr. Julian Kenyon Beechwood, Embley Lane East Wellow, Near Romsey, Hampshire, SO51 6DN, United Kingdom (3) | 10,812,064 | 13.11 | % | ||||||
| Common Stock | All directors and executive officers as a group | 20,513,825 | 24.88 | % | ||||||
| 5% Shareholders: | ||||||||||
| Common Stock | Ostrowski Properties Pty Ltd. 33 Allambee Avenue Elsternwick, VIC, 3185, Australia (4) | 4,132,691 | 5.01 | % | ||||||
| Common Stock | Putney Consultants Ltd. (5) | 32,938,614 | 39.95 | % | ||||||
| 44 |
| (1) | Applicable percentages are based on 82,444,100 shares outstanding, adjusted as required by rules of the SEC. Beneficial ownership is determined under the rules of the SEC and generally includes voting or investment power with respect to securities. Shares of common stock subject to options, warrants and convertible notes currently exercisable or convertible, or exercisable or convertible within 60 days are deemed outstanding for computing the percentage of the person holding such securities but are not deemed outstanding for computing the percentage of any other person. Unless otherwise indicated in the footnotes to this table, Propanc believes that each of the shareholders named in the table has sole voting and investment power with respect to the shares of common stock indicated as beneficially owned by them. |
| (2) | Mr. Nathanielsz is a director and executive officer. Represents shares of common stock held by North Horizon Investments Pty Ltd., a Nathanielsz Family Trust. Mr. Nathanielsz has voting and investment power over these shares. |
| (3) | Dr. Kenyon is a director. Represents shares of common stock. |
| (4) | Mr. Jan Ostrowski and Mrs. Ywonna Ostrowski, Mr. Nathanielsz's father-in-law and mother-in-law, have voting power and investment power of Ostrowski Properties PTY Ltd. |
| (5) | Dr. Douglas Mitchell, a former director and executive officer of the Company, has voting and investment power of Putney Consultants Ltd. |
Item 13. Certain Relationships and Related Transactions, and Director Independence.
Certain Relationships and Related Transactions
Since inception, Propanc Health Group Corporation has conducted transactions with directors and director related entities. These transactions included the following:
As of June 30, 2014 and 2013, the Company owed certain directors a total of $161,975 and $130,689 respectively, for money loaned to the Company throughout the years. The loan balance owed at June 30, 2014 was not interest bearing.
As of June 30, 2014 and 2013, the Company owed certain current and former directors a total of $60,350 and $57,237, respectively, related to expenses incurred on behalf of the Company related to corporate startup costs and intellectual property.
| Aggregate Amount of Principal Loaned | Amount of Principal Paid | Outstanding Balance as of June 30 , | ||||||||||||||||||||||||
| Name of Related Party | Affiliation | 2014 | 2013 | 2014 | 2013 | 2014 | 2013 | |||||||||||||||||||
| James Nathanielsz | Chief Executive Officer & Chairman of the Board | $ | 86,225 | * | $ | 80,272 | * | $ | 59,084 | * | $ | 30,438 | * | $ | 78,586 | * | $ | 49,835 | * | |||||||
| Julian Kenyon | Director | $ | - | $ | 13,269 | $ | - | $ | - | $ | 13,686 | * | $ | 13,269 | * | |||||||||||
| Douglas Mitchell | Former Chief Executive Officer and Chairman of the Board | $ | - | $ | - | $ | - | $ | - | $ | 69,703 | * | $ | 67,584 | * | |||||||||||
| 45 |
* Loans were made in Australian Dollar and the difference of amount is due to the fluctuating currency exchange rate between U.S. Dollar and Australian Dollar.
The loans of $161,975 and $130,689 as of June 30, 2014 and 2013, respectively, are reflected as stockholder loans in the consolidated balance sheets.
Item 14. Principal Accounting Fees and Services.
The Company's board of directors reviews and approves audit and permissible non-audit services performed by its independent registered public accounting firm, as well as the fees charged for such services. In its review of non-audit service and its appointment of Salberg & Company, P.A. as our independent registered public accounting firm, the board considered whether the provision of such services is compatible with maintaining independence. All of the services provided and fees charged by Salberg & Company, P.A. in 2014 and 2013 were approved by the board of directors. The following table shows the fees for the years ended June 30, 2014 and 2013:
| 2014 | 2013 | |||||||
| Audit Fees (1) | $ | 40,200 | $ | 38,000 | ||||
| Audit Related Fees (2) | $ | 650 | $ | 3,300 | ||||
| Tax Fees (3) | $ | - | $ | - | ||||
| All Other Fees | $ | - | $ | - | ||||
| Total | $ | 40,850 | $ | 41,300 | ||||
(1) Audit fees – these fees relate to the audit of our annual consolidated financial statements and the review of our interim quarterly consolidated financial statements.
(2) Audit related fees – these fees relate primarily to the auditors’ review of our registration statements and audit related consulting.
(3) Tax fees – no fees of this sort were billed by Salberg & Company P.A., our principal accountant during 2014 and 2013.
All Other Fees
We did not incur any other fees related to services rendered by our independent registered public accounting firm for the fiscal years ended June 30, 2014 and 2013.
The SEC requires that before our independent registered public accounting firm is engaged by us to render any auditing or permitted non-audit related service, the engagement be either: (i) approved by our audit committee or (ii) entered into pursuant to pre-approval policies and procedures established by the audit committee, provided that the policies and procedures are detailed as to the particular service, the audit committee is informed of each service, and such policies and procedures do not include delegation of the audit committee’s responsibilities to management.
We do not have an audit committee. Our Board pre-approves all services provided by our independent registered public accounting firm. All of the above services and fees during 2014 and 2013 were reviewed and approved by our Board of Directors either before or after the respective services were rendered.
| 46 |
(a) Exhibits
| Exhibit Number | Description | |
| 3.1 | Articles of Incorporation, incorporated by reference to Exhibit 3.1 to the Company’s Registration Statement on Form S-1, as amended; filed with the SEC on June 23, 2011. | |
| 3.2 | Bylaws, incorporated by reference to Exhibit 3.2 to the Company’s Registration Statement on Form S-1, as amended; filed with the SEC on June 23, 2011. | |
| 4.1 | Form of 8% Convertible Redeemable Note issued to Union Capital LLC (“Union”) by the Company dated May 30, 2014, incorporated by reference to Exhibit 4.1 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 4.2 | Form of 8% Convertible Redeemable Back End Note issued to Union by the Company dated May 30, 2014, incorporated by reference to Exhibit 4.2 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 4.3 | Form of 8% Collateralized Secured Promissory note Back End Note issued to the Company by Union dated May 30, 2014, incorporated by reference to Exhibit 4.3 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 4.4 | Form of 8% Convertible Redeemable Note issued to LG Capital Funding (“LG”) by the Company dated May 29, 2014, incorporated by reference to Exhibit 4.4 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 4.5 | Form of 8% Convertible Redeemable Back End Note issued to LG by the Company dated May 29, 2014, incorporated by reference to Exhibit 4.5 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 4.6 | Form of 8% Collateralized Secured Promissory note Back End Note issued to the Company by LG dated May 29, 2014, incorporated by reference to Exhibit 4.6 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 4.7 | Form of 8% Convertible Redeemable Note issued to Adar Bays, LLC (“Adar”) by the Company dated May 29, 2014, incorporated by reference to Exhibit 4.7 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 4.8 | Form of 8% Convertible Redeemable Back End Note issued to Adar by the Company dated May 29, 2014, incorporated by reference to Exhibit 4.8 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 4.9 | Form of 8% Collateralized Secured Promissory note Back End Note issued to the Company by Adar dated May 29, 2014, incorporated by reference to Exhibit 4.9 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 4.10 | 10% Convertible Note issued to Tarpon Bay Partners LLC (“Tarpon”) dated May 8, 2014. | |
| 4.11 | Promissory Note issued to Southridge Partners II, L.P. (Southridge) dated July 17, 2014. | |
| 10.1 | Exchange Offer Term Sheet, incorporated by reference to Exhibit 10.2 to Company’s Registration Statement on Form S-1, as amended; filed with the Securities and Exchange Commission on June 23, 2011. | |
| 10.2 | Exchange Offer Registration Rights Agreement, incorporated by reference to Exhibit 10.3 to Company’s Registration Statement on Form S-1, as amended; filed with the Securities and Exchange Commission on June 23, 2011. | |
| 10.3 | Exchange Offer Subscription Agreement, incorporated by reference to Exhibit 10.3 to Company’s Registration Statement on Form S-1, as amended; filed with the Securities and Exchange Commission on June 23, 2011. | |
| 10.4 | Employment Agreement dated August 15, 2010 between the Company and James Nathanielsz, incorporated by reference to Exhibit 10.1 to registrant’s Registration Statement on Form S-1, as amended; filed with the Securities and Exchange Commission on June 23, 2011. | |
| 10.5 | Form of Form of Securities Purchase Agreement between the Company and Union dated May 30, 2014, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 10.6 | Form of Securities Purchase Agreement between the Company and LG dated May 29, 2014, incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 10.7 | Form of Securities Purchase Agreement between the Company and Adar dated May 29, 2014, incorporated by reference to Exhibit 10.3 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 10.8 | Settlement Agreement and Stipulation between the Company and Tarpon dated July 18, 2014, incorporated by reference to Exhibit 10.4 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 10.9 | Order Granting Approval of Settlement Agreement and Stipulation between the Company and Tarpon dated September 9, 2014, incorporated by reference to Exhibit 10.5 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 10.10 | Form of Equity Purchase Agreement between the Company and Southridge Partners II L.P. (“Southridge”), incorporated by reference to Exhibit 10.6 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 10.11 | Form of Registration Rights Agreement between Company and Southridge, incorporated by reference to Exhibit 10.7 to the Current Report on Form 8-K filed on September 23, 2014. | |
| 31.1* | Certifications of the Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
| 32.1+ | Certification Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |
| 101. INS** | XBRL Instance Document. | |
| 101. SCH** | XBRL Taxonomy Extension Schema Document | |
| 101. CAL** | XBRL Taxonomy Extension Calculation Linkbase Document. | |
| 101. LAB** | XBRL Taxonomy Extension Label Linkbase Document. | |
| 101. PRE** | XBRL Taxonomy Extension Presentation Linkbase Document. | |
| 101. DEF** | XBRL Taxonomy Extension Definition Linkbase Document. |
| * | Filed herewith |
| + | Furnished herewith |
| ** | Furnished and not filed or a part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act of 1933, as amended, deemed not filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, and otherwise not subject to liability under these sections. |
| 47 |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| PROPANC HEALTH GROUP CORPORATION | ||
| Dated: October 14, 2014 | By: | /s/ James Nathanielsz |
| James Nathanielsz | ||
| Chief Executive Officer (Principal Executive Officer) | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Name | Title | Date | ||
| /s/ James Nathanielsz | Chief Executive Officer and | October 14, 2014 | ||
| James Nathanielsz | Director (Principal Executive Officer) | |||
| /s/ Julian Kenyon | ||||
| Julian Kenyon | Director | October 14, 2014 |
| 48 |